 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepEra: A Deep Evidence Reranking Agent for Scientific Retrieval-Augmented Generated Question Answering
Jan 23, 2026With the rapid growth of scientific literature, scientific question answering (SciQA) has become increasingly critical for exploring and utilizing scientific knowledge. Retrieval-Augmented Generation (RAG) enhances LLMs by incorporating knowledge from external sources, thereby providing credible evidence for scientific question answering. But existing retrieval and reranking methods remain vulnerable to passages that are semantically similar but logically irrelevant, often reducing factual reliability and amplifying hallucinations.To address this challenge, we propose a Deep Evidence Reranking Agent (DeepEra) that integrates step-by-step reasoning, enabling more precise evaluation of candidate passages beyond surface-level semantics. To support systematic evaluation, we construct SciRAG-SSLI (Scientific RAG - Semantically Similar but Logically Irrelevant), a large-scale dataset comprising about 300K SciQA instances across 10 subjects, constructed from 10M scientific corpus. The dataset combines naturally retrieved contexts with systematically generated distractors to test logical robustness and factual grounding. Comprehensive evaluations confirm that our approach achieves superior retrieval performance compared to leading rerankers. To our knowledge, this work is the first to comprehensively study and empirically validate innegligible SSLI issues in two-stage RAG frameworks.
SciHorizon-GENE: Benchmarking LLM for Life Sciences Inference from Gene Knowledge to Functional Understanding
Jan 21, 2026Large language models (LLMs) have shown growing promise in biomedical research, particularly for knowledge-driven interpretation tasks. However, their ability to reliably reason from gene-level knowledge to functional understanding, a core requirement for knowledge-enhanced cell atlas interpretation, remains largely underexplored. To address this gap, we introduce SciHorizon-GENE, a large-scale gene-centric benchmark constructed from authoritative biological databases. The benchmark integrates curated knowledge for over 190K human genes and comprises more than 540K questions covering diverse gene-to-function reasoning scenarios relevant to cell type annotation, functional interpretation, and mechanism-oriented analysis. Motivated by behavioral patterns observed in preliminary examinations, SciHorizon-GENE evaluates LLMs along four biologically critical perspectives: research attention sensitivity, hallucination tendency, answer completeness, and literature influence, explicitly targeting failure modes that limit the safe adoption of LLMs in biological interpretation pipelines. We systematically evaluate a wide range of state-of-the-art general-purpose and biomedical LLMs, revealing substantial heterogeneity in gene-level reasoning capabilities and persistent challenges in generating faithful, complete, and literature-grounded functional interpretations. Our benchmark establishes a systematic foundation for analyzing LLM behavior at the gene scale and offers insights for model selection and development, with direct relevance to knowledge-enhanced biological interpretation.
ScienceDB AI: An LLM-Driven Agentic Recommender System for Large-Scale Scientific Data Sharing Services
Jan 03, 2026The rapid growth of AI for Science (AI4S) has underscored the significance of scientific datasets, leading to the establishment of numerous national scientific data centers and sharing platforms. Despite this progress, efficiently promoting dataset sharing and utilization for scientific research remains challenging. Scientific datasets contain intricate domain-specific knowledge and contexts, rendering traditional collaborative filtering-based recommenders inadequate. Recent advances in Large Language Models (LLMs) offer unprecedented opportunities to build conversational agents capable of deep semantic understanding and personalized recommendations. In response, we present ScienceDB AI, a novel LLM-driven agentic recommender system developed on Science Data Bank (ScienceDB), one of the largest global scientific data-sharing platforms. ScienceDB AI leverages natural language conversations and deep reasoning to accurately recommend datasets aligned with researchers' scientific intents and evolving requirements. The system introduces several innovations: a Scientific Intention Perceptor to extract structured experimental elements from complicated queries, a Structured Memory Compressor to manage multi-turn dialogues effectively, and a Trustworthy Retrieval-Augmented Generation (Trustworthy RAG) framework. The Trustworthy RAG employs a two-stage retrieval mechanism and provides citable dataset references via Citable Scientific Task Record (CSTR) identifiers, enhancing recommendation trustworthiness and reproducibility. Through extensive offline and online experiments using over 10 million real-world datasets, ScienceDB AI has demonstrated significant effectiveness. To our knowledge, ScienceDB AI is the first LLM-driven conversational recommender tailored explicitly for large-scale scientific dataset sharing services. The platform is publicly accessible at: https://ai.scidb.cn/en.
SciRerankBench: Benchmarking Rerankers Towards Scientific Retrieval-Augmented Generated LLMs
Aug 12, 2025Scientific literature question answering is a pivotal step towards new scientific discoveries. Recently, \textit{two-stage} retrieval-augmented generated large language models (RAG-LLMs) have shown impressive advancements in this domain. Such a two-stage framework, especially the second stage (reranker), is particularly essential in the scientific domain, where subtle differences in terminology may have a greatly negative impact on the final factual-oriented or knowledge-intensive answers. Despite this significant progress, the potential and limitations of these works remain unexplored. In this work, we present a Scientific Rerank-oriented RAG Benchmark (SciRerankBench), for evaluating rerankers within RAG-LLMs systems, spanning five scientific subjects. To rigorously assess the reranker performance in terms of noise resilience, relevance disambiguation, and factual consistency, we develop three types of question-context-answer (Q-C-A) pairs, i.e., Noisy Contexts (NC), Semantically Similar but Logically Irrelevant Contexts (SSLI), and Counterfactual Contexts (CC). Through systematic evaluation of 13 widely used rerankers on five families of LLMs, we provide detailed insights into their relative strengths and limitations. To the best of our knowledge, SciRerankBench is the first benchmark specifically developed to evaluate rerankers within RAG-LLMs, which provides valuable observations and guidance for their future development.
QUITE: A Query Rewrite System Beyond Rules with LLM Agents
Jun 09, 2025Query rewrite transforms SQL queries into semantically equivalent forms that run more efficiently. Existing approaches mainly rely on predefined rewrite rules, but they handle a limited subset of queries and can cause performance regressions. This limitation stems from three challenges of rule-based query rewrite: (1) it is hard to discover and verify new rules, (2) fixed rewrite rules do not generalize to new query patterns, and (3) some rewrite techniques cannot be expressed as fixed rules. Motivated by the fact that human experts exhibit significantly better rewrite ability but suffer from scalability, and Large Language Models (LLMs) have demonstrated nearly human-level semantic and reasoning abilities, we propose a new approach of using LLMs to rewrite SQL queries beyond rules. Due to the hallucination problems in LLMs, directly applying LLMs often leads to nonequivalent and suboptimal queries. To address this issue, we propose QUITE (query rewrite), a training-free and feedback-aware system based on LLM agents that rewrites SQL queries into semantically equivalent forms with significantly better performance, covering a broader range of query patterns and rewrite strategies compared to rule-based methods. Firstly, we design a multi-agent framework controlled by a finite state machine (FSM) to equip LLMs with the ability to use external tools and enhance the rewrite process with real-time database feedback. Secondly, we develop a rewrite middleware to enhance the ability of LLMs to generate optimized query equivalents. Finally, we employ a novel hint injection technique to improve execution plans for rewritten queries. Extensive experiments show that QUITE reduces query execution time by up to 35.8% over state-of-the-art approaches and produces 24.1% more rewrites than prior methods, covering query cases that earlier systems did not handle.
Distilling Closed-Source LLM's Knowledge for Locally Stable and Economic Biomedical Entity Linking
May 26, 2025Biomedical entity linking aims to map nonstandard entities to standard entities in a knowledge base. Traditional supervised methods perform well but require extensive annotated data to transfer, limiting their usage in low-resource scenarios. Large language models (LLMs), especially closed-source LLMs, can address these but risk stability issues and high economic costs: using these models is restricted by commercial companies and brings significant economic costs when dealing with large amounts of data. To address this, we propose ``RPDR'', a framework combining closed-source LLMs and open-source LLMs for re-ranking candidates retrieved by a retriever fine-tuned with a small amount of data. By prompting a closed-source LLM to generate training data from unannotated data and fine-tuning an open-source LLM for re-ranking, we effectively distill the knowledge to the open-source LLM that can be deployed locally, thus avoiding the stability issues and the problem of high economic costs. We evaluate RPDR on two datasets, including one real-world dataset and one publicly available dataset involving two languages: Chinese and English. RPDR achieves 0.019 Acc@1 improvement and 0.036 Acc@1 improvement on the Aier dataset and the Ask A Patient dataset when the amount of training data is not enough. The results demonstrate the superiority and generalizability of the proposed framework.
Disentangled Multi-span Evolutionary Network against Temporal Knowledge Graph Reasoning
May 20, 2025Temporal Knowledge Graphs (TKGs), as an extension of static Knowledge Graphs (KGs), incorporate the temporal feature to express the transience of knowledge by describing when facts occur. TKG extrapolation aims to infer possible future facts based on known history, which has garnered significant attention in recent years. Some existing methods treat TKG as a sequence of independent subgraphs to model temporal evolution patterns, demonstrating impressive reasoning performance. However, they still have limitations: 1) In modeling subgraph semantic evolution, they usually neglect the internal structural interactions between subgraphs, which are actually crucial for encoding TKGs. 2) They overlook the potential smooth features that do not lead to semantic changes, which should be distinguished from the semantic evolution process. Therefore, we propose a novel Disentangled Multi-span Evolutionary Network (DiMNet) for TKG reasoning. Specifically, we design a multi-span evolution strategy that captures local neighbor features while perceiving historical neighbor semantic information, thus enabling internal interactions between subgraphs during the evolution process. To maximize the capture of semantic change patterns, we design a disentangle component that adaptively separates nodes' active and stable features, used to dynamically control the influence of historical semantics on future evolution. Extensive experiments conducted on four real-world TKG datasets show that DiMNet demonstrates substantial performance in TKG reasoning, and outperforms the state-of-the-art up to 22.7% in MRR.
scSiameseClu: A Siamese Clustering Framework for Interpreting single-cell RNA Sequencing Data
May 19, 2025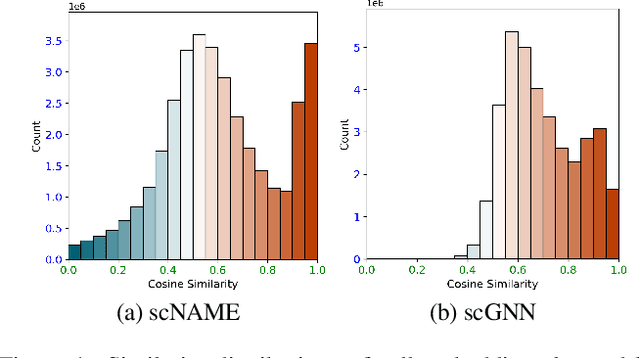
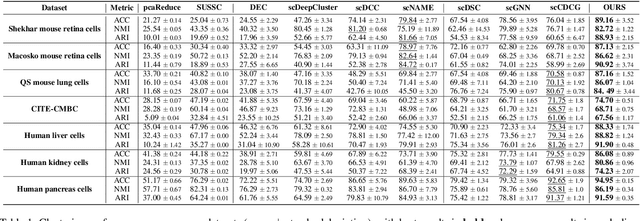
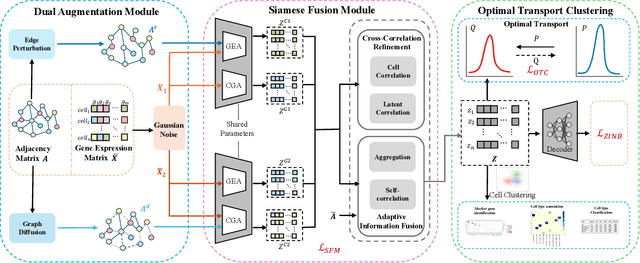
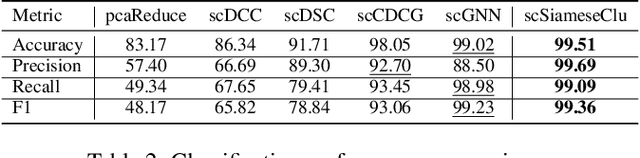
Single-cell RNA sequencing (scRNA-seq) reveals cell heterogeneity, with cell clustering playing a key role in identifying cell types and marker genes. Recent advances, especially graph neural networks (GNNs)-based methods, have significantly improved clustering performance. However, the analysis of scRNA-seq data remains challenging due to noise, sparsity, and high dimensionality. Compounding these challenges, GNNs often suffer from over-smoothing, limiting their ability to capture complex biological information. In response, we propose scSiameseClu, a novel Siamese Clustering framework for interpreting single-cell RNA-seq data, comprising of 3 key steps: (1) Dual Augmentation Module, which applies biologically informed perturbations to the gene expression matrix and cell graph relationships to enhance representation robustness; (2) Siamese Fusion Module, which combines cross-correlation refinement and adaptive information fusion to capture complex cellular relationships while mitigating over-smoothing; and (3) Optimal Transport Clustering, which utilizes Sinkhorn distance to efficiently align cluster assignments with predefined proportions while maintaining balance. Comprehensive evaluations on seven real-world datasets demonstrate that~\methodname~outperforms state-of-the-art methods in single-cell clustering, cell type annotation, and cell type classification, providing a powerful tool for scRNA-seq data interpretation.
Rethinking Graph Contrastive Learning through Relative Similarity Preservation
May 08, 2025Graph contrastive learning (GCL) has achieved remarkable success by following the computer vision paradigm of preserving absolute similarity between augmented views. However, this approach faces fundamental challenges in graphs due to their discrete, non-Euclidean nature -- view generation often breaks semantic validity and similarity verification becomes unreliable. Through analyzing 11 real-world graphs, we discover a universal pattern transcending the homophily-heterophily dichotomy: label consistency systematically diminishes as structural distance increases, manifesting as smooth decay in homophily graphs and oscillatory decay in heterophily graphs. We establish theoretical guarantees for this pattern through random walk theory, proving label distribution convergence and characterizing the mechanisms behind different decay behaviors. This discovery reveals that graphs naturally encode relative similarity patterns, where structurally closer nodes exhibit collectively stronger semantic relationships. Leveraging this insight, we propose RELGCL, a novel GCL framework with complementary pairwise and listwise implementations that preserve these inherent patterns through collective similarity objectives. Extensive experiments demonstrate that our method consistently outperforms 20 existing approaches across both homophily and heterophily graphs, validating the effectiveness of leveraging natural relative similarity over artificial absolute similarity.
m-KAILIN: Knowledge-Driven Agentic Scientific Corpus Distillation Framework for Biomedical Large Language Models Training
Apr 28, 2025The rapid progress of large language models (LLMs) in biomedical research has underscored the limitations of existing open-source annotated scientific corpora, which are often insufficient in quantity and quality. Addressing the challenge posed by the complex hierarchy of biomedical knowledge, we propose a knowledge-driven, multi-agent framework for scientific corpus distillation tailored for LLM training in the biomedical domain. Central to our approach is a collaborative multi-agent architecture, where specialized agents, each guided by the Medical Subject Headings (MeSH) hierarchy, work in concert to autonomously extract, synthesize, and self-evaluate high-quality textual data from vast scientific literature. These agents collectively generate and refine domain-specific question-answer pairs, ensuring comprehensive coverage and consistency with biomedical ontologies while minimizing manual involvement. Extensive experimental results show that language models trained on our multi-agent distilled datasets achieve notable improvements in biomedical question-answering tasks, outperforming both strong life sciences LLM baselines and advanced proprietary models. Notably, our AI-Ready dataset enables Llama3-70B to surpass GPT-4 with MedPrompt and Med-PaLM-2, despite their larger scale. Detailed ablation studies and case analyses further validate the effectiveness and synergy of each agent within the framework, highlighting the potential of multi-agent collaboration in biomedical LLM training.