 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeepEra: A Deep Evidence Reranking Agent for Scientific Retrieval-Augmented Generated Question Answering
Jan 23, 2026With the rapid growth of scientific literature, scientific question answering (SciQA) has become increasingly critical for exploring and utilizing scientific knowledge. Retrieval-Augmented Generation (RAG) enhances LLMs by incorporating knowledge from external sources, thereby providing credible evidence for scientific question answering. But existing retrieval and reranking methods remain vulnerable to passages that are semantically similar but logically irrelevant, often reducing factual reliability and amplifying hallucinations.To address this challenge, we propose a Deep Evidence Reranking Agent (DeepEra) that integrates step-by-step reasoning, enabling more precise evaluation of candidate passages beyond surface-level semantics. To support systematic evaluation, we construct SciRAG-SSLI (Scientific RAG - Semantically Similar but Logically Irrelevant), a large-scale dataset comprising about 300K SciQA instances across 10 subjects, constructed from 10M scientific corpus. The dataset combines naturally retrieved contexts with systematically generated distractors to test logical robustness and factual grounding. Comprehensive evaluations confirm that our approach achieves superior retrieval performance compared to leading rerankers. To our knowledge, this work is the first to comprehensively study and empirically validate innegligible SSLI issues in two-stage RAG frameworks.
SciHorizon-GENE: Benchmarking LLM for Life Sciences Inference from Gene Knowledge to Functional Understanding
Jan 21, 2026Large language models (LLMs) have shown growing promise in biomedical research, particularly for knowledge-driven interpretation tasks. However, their ability to reliably reason from gene-level knowledge to functional understanding, a core requirement for knowledge-enhanced cell atlas interpretation, remains largely underexplored. To address this gap, we introduce SciHorizon-GENE, a large-scale gene-centric benchmark constructed from authoritative biological databases. The benchmark integrates curated knowledge for over 190K human genes and comprises more than 540K questions covering diverse gene-to-function reasoning scenarios relevant to cell type annotation, functional interpretation, and mechanism-oriented analysis. Motivated by behavioral patterns observed in preliminary examinations, SciHorizon-GENE evaluates LLMs along four biologically critical perspectives: research attention sensitivity, hallucination tendency, answer completeness, and literature influence, explicitly targeting failure modes that limit the safe adoption of LLMs in biological interpretation pipelines. We systematically evaluate a wide range of state-of-the-art general-purpose and biomedical LLMs, revealing substantial heterogeneity in gene-level reasoning capabilities and persistent challenges in generating faithful, complete, and literature-grounded functional interpretations. Our benchmark establishes a systematic foundation for analyzing LLM behavior at the gene scale and offers insights for model selection and development, with direct relevance to knowledge-enhanced biological interpretation.
ScienceDB AI: An LLM-Driven Agentic Recommender System for Large-Scale Scientific Data Sharing Services
Jan 03, 2026The rapid growth of AI for Science (AI4S) has underscored the significance of scientific datasets, leading to the establishment of numerous national scientific data centers and sharing platforms. Despite this progress, efficiently promoting dataset sharing and utilization for scientific research remains challenging. Scientific datasets contain intricate domain-specific knowledge and contexts, rendering traditional collaborative filtering-based recommenders inadequate. Recent advances in Large Language Models (LLMs) offer unprecedented opportunities to build conversational agents capable of deep semantic understanding and personalized recommendations. In response, we present ScienceDB AI, a novel LLM-driven agentic recommender system developed on Science Data Bank (ScienceDB), one of the largest global scientific data-sharing platforms. ScienceDB AI leverages natural language conversations and deep reasoning to accurately recommend datasets aligned with researchers' scientific intents and evolving requirements. The system introduces several innovations: a Scientific Intention Perceptor to extract structured experimental elements from complicated queries, a Structured Memory Compressor to manage multi-turn dialogues effectively, and a Trustworthy Retrieval-Augmented Generation (Trustworthy RAG) framework. The Trustworthy RAG employs a two-stage retrieval mechanism and provides citable dataset references via Citable Scientific Task Record (CSTR) identifiers, enhancing recommendation trustworthiness and reproducibility. Through extensive offline and online experiments using over 10 million real-world datasets, ScienceDB AI has demonstrated significant effectiveness. To our knowledge, ScienceDB AI is the first LLM-driven conversational recommender tailored explicitly for large-scale scientific dataset sharing services. The platform is publicly accessible at: https://ai.scidb.cn/en.
SciRerankBench: Benchmarking Rerankers Towards Scientific Retrieval-Augmented Generated LLMs
Aug 12, 2025Scientific literature question answering is a pivotal step towards new scientific discoveries. Recently, \textit{two-stage} retrieval-augmented generated large language models (RAG-LLMs) have shown impressive advancements in this domain. Such a two-stage framework, especially the second stage (reranker), is particularly essential in the scientific domain, where subtle differences in terminology may have a greatly negative impact on the final factual-oriented or knowledge-intensive answers. Despite this significant progress, the potential and limitations of these works remain unexplored. In this work, we present a Scientific Rerank-oriented RAG Benchmark (SciRerankBench), for evaluating rerankers within RAG-LLMs systems, spanning five scientific subjects. To rigorously assess the reranker performance in terms of noise resilience, relevance disambiguation, and factual consistency, we develop three types of question-context-answer (Q-C-A) pairs, i.e., Noisy Contexts (NC), Semantically Similar but Logically Irrelevant Contexts (SSLI), and Counterfactual Contexts (CC). Through systematic evaluation of 13 widely used rerankers on five families of LLMs, we provide detailed insights into their relative strengths and limitations. To the best of our knowledge, SciRerankBench is the first benchmark specifically developed to evaluate rerankers within RAG-LLMs, which provides valuable observations and guidance for their future development.
Collaborative Multi-Agent Reinforcement Learning for Automated Feature Transformation with Graph-Driven Path Optimization
Apr 24, 2025



Feature transformation methods aim to find an optimal mathematical feature-feature crossing process that generates high-value features and improves the performance of downstream machine learning tasks. Existing frameworks, though designed to mitigate manual costs, often treat feature transformations as isolated operations, ignoring dynamic dependencies between transformation steps. To address the limitations, we propose TCTO, a collaborative multi-agent reinforcement learning framework that automates feature engineering through graph-driven path optimization. The framework's core innovation lies in an evolving interaction graph that models features as nodes and transformations as edges. Through graph pruning and backtracking, it dynamically eliminates low-impact edges, reduces redundant operations, and enhances exploration stability. This graph also provides full traceability to empower TCTO to reuse high-utility subgraphs from historical transformations. To demonstrate the efficacy and adaptability of our approach, we conduct comprehensive experiments and case studies, which show superior performance across a range of datasets.
Comprehend, Divide, and Conquer: Feature Subspace Exploration via Multi-Agent Hierarchical Reinforcement Learning
Apr 24, 2025



Feature selection aims to preprocess the target dataset, find an optimal and most streamlined feature subset, and enhance the downstream machine learning task. Among filter, wrapper, and embedded-based approaches, the reinforcement learning (RL)-based subspace exploration strategy provides a novel objective optimization-directed perspective and promising performance. Nevertheless, even with improved performance, current reinforcement learning approaches face challenges similar to conventional methods when dealing with complex datasets. These challenges stem from the inefficient paradigm of using one agent per feature and the inherent complexities present in the datasets. This observation motivates us to investigate and address the above issue and propose a novel approach, namely HRLFS. Our methodology initially employs a Large Language Model (LLM)-based hybrid state extractor to capture each feature's mathematical and semantic characteristics. Based on this information, features are clustered, facilitating the construction of hierarchical agents for each cluster and sub-cluster. Extensive experiments demonstrate the efficiency, scalability, and robustness of our approach. Compared to contemporary or the one-feature-one-agent RL-based approaches, HRLFS improves the downstream ML performance with iterative feature subspace exploration while accelerating total run time by reducing the number of agents involved.
Large Language Model Agent: A Survey on Methodology, Applications and Challenges
Mar 27, 2025The era of intelligent agents is upon us, driven by revolutionary advancements in large language models. Large Language Model (LLM) agents, with goal-driven behaviors and dynamic adaptation capabilities, potentially represent a critical pathway toward artificial general intelligence. This survey systematically deconstructs LLM agent systems through a methodology-centered taxonomy, linking architectural foundations, collaboration mechanisms, and evolutionary pathways. We unify fragmented research threads by revealing fundamental connections between agent design principles and their emergent behaviors in complex environments. Our work provides a unified architectural perspective, examining how agents are constructed, how they collaborate, and how they evolve over time, while also addressing evaluation methodologies, tool applications, practical challenges, and diverse application domains. By surveying the latest developments in this rapidly evolving field, we offer researchers a structured taxonomy for understanding LLM agents and identify promising directions for future research. The collection is available at https://github.com/luo-junyu/Awesome-Agent-Papers.
FastFT: Accelerating Reinforced Feature Transformation via Advanced Exploration Strategies
Mar 26, 2025



Feature Transformation is crucial for classic machine learning that aims to generate feature combinations to enhance the performance of downstream tasks from a data-centric perspective. Current methodologies, such as manual expert-driven processes, iterative-feedback techniques, and exploration-generative tactics, have shown promise in automating such data engineering workflow by minimizing human involvement. However, three challenges remain in those frameworks: (1) It predominantly depends on downstream task performance metrics, as assessment is time-consuming, especially for large datasets. (2) The diversity of feature combinations will hardly be guaranteed after random exploration ends. (3) Rare significant transformations lead to sparse valuable feedback that hinders the learning processes or leads to less effective results. In response to these challenges, we introduce FastFT, an innovative framework that leverages a trio of advanced strategies.We first decouple the feature transformation evaluation from the outcomes of the generated datasets via the performance predictor. To address the issue of reward sparsity, we developed a method to evaluate the novelty of generated transformation sequences. Incorporating this novelty into the reward function accelerates the model's exploration of effective transformations, thereby improving the search productivity. Additionally, we combine novelty and performance to create a prioritized memory buffer, ensuring that essential experiences are effectively revisited during exploration. Our extensive experimental evaluations validate the performance, efficiency, and traceability of our proposed framework, showcasing its superiority in handling complex feature transformation tasks.
Knowledge Hierarchy Guided Biological-Medical Dataset Distillation for Domain LLM Training
Jan 25, 2025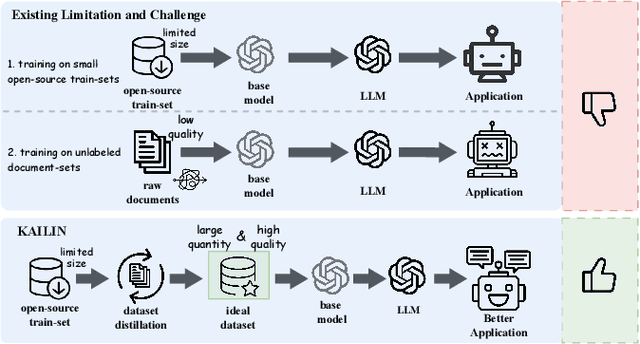

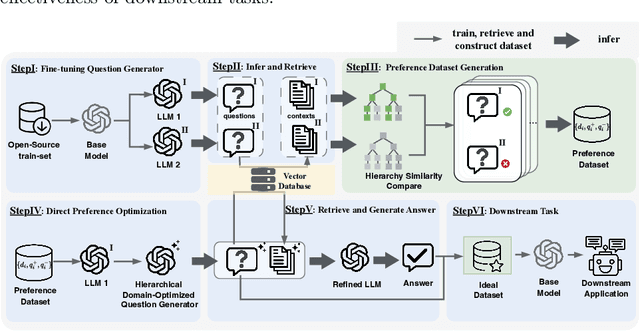

The rapid advancement of large language models (LLMs) in biological-medical applications has highlighted a gap between their potential and the limited scale and often low quality of available open-source annotated textual datasets. In addition, the inherent complexity of the biomedical knowledge hierarchy significantly hampers efforts to bridge this gap.Can LLMs themselves play a pivotal role in overcoming this limitation? Motivated by this question, we investigate this challenge in the present study.We propose a framework that automates the distillation of high-quality textual training data from the extensive scientific literature. Our approach self-evaluates and generates questions that are more closely aligned with the biomedical domain, guided by the biomedical knowledge hierarchy through medical subject headings (MeSH). This comprehensive framework establishes an automated workflow, thereby eliminating the need for manual intervention. Furthermore, we conducted comprehensive experiments to evaluate the impact of our framework-generated data on downstream language models of varying sizes. Our approach substantially improves question-answering tasks compared to pre-trained models from the life sciences domain and powerful close-source models represented by GPT-4. Notably, the generated AI-Ready dataset enabled the Llama3-70B base model to outperform GPT-4 using MedPrompt with multiple times the number of parameters. Detailed case studies and ablation experiments underscore the significance of each component within our framework
Comprehensive Metapath-based Heterogeneous Graph Transformer for Gene-Disease Association Prediction
Jan 14, 2025



Discovering gene-disease associations is crucial for understanding disease mechanisms, yet identifying these associations remains challenging due to the time and cost of biological experiments. Computational methods are increasingly vital for efficient and scalable gene-disease association prediction. Graph-based learning models, which leverage node features and network relationships, are commonly employed for biomolecular predictions. However, existing methods often struggle to effectively integrate node features, heterogeneous structures, and semantic information. To address these challenges, we propose COmprehensive MEtapath-based heterogeneous graph Transformer(COMET) for predicting gene-disease associations. COMET integrates diverse datasets to construct comprehensive heterogeneous networks, initializing node features with BioGPT. We define seven Metapaths and utilize a transformer framework to aggregate Metapath instances, capturing global contexts and long-distance dependencies. Through intra- and inter-metapath aggregation using attention mechanisms, COMET fuses latent vectors from multiple Metapaths to enhance GDA prediction accuracy. Our method demonstrates superior robustness compared to state-of-the-art approaches. Ablation studies and visualizations validate COMET's effectiveness, providing valuable insights for advancing human health research.