 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Serving for Dynamic Agent Workflows with Prediction-based KV-Cache Management
May 07, 2026LLM-based workflows compose specialized agents to execute complex tasks, and these agents usually share substantial context, allowing KV-Cache reuse to save computation. Existing approaches either manage KV-Cache at agent level and fail to exploit the reuse opportunities within workflows, or manage cache at the workflow level but assume that each workflow calls a static sequence of agents. However, practical workflows are typically dynamic, where the sequence of invoked agents and thus induced cache reuse opportunities depend on the context of each task. To serve such dynamic workflows efficiently, we build a system dubbed PBKV (\textbf{P}rediction-\textbf{B}ased \textbf{KV}-Cache Management). For each workflow, PBKV predicts the agent invocations in several future steps by fusing the guidance from historical workflows and context of the target workflow. Based on the predictions, PBKV estimates the reuse potential of cache entries and keeps the high-potential entries in GPU memory. To be robust to prediction errors, PBKV utilizes the predictions conservatively during both cache eviction and prefetching. Experiments on three workflow benchmarks show that PBKV achieves up to $1.85\times$ speedup over LRU on dynamic workflows, and up to $1.26\times$ speedup over the SOTA baseline KVFlow on the static workflow.
Projection Guided Personalized Federated Learning for Low Dose CT Denoising
Mar 12, 2026Low-dose CT (LDCT) reduces radiation exposure but introduces protocol-dependent noise and artifacts that vary across institutions. While federated learning enables collaborative training without centralizing patient data, existing methods personalize in image space, making it difficult to separate scanner noise from patient anatomy. We propose ProFed (Projection Guided Personalized Federated Learning), a framework that complements the image space approach by performing dual-level personalization in the projection space, where noise originates during CT measurements before reconstruction combines protocol and anatomy effects. ProFed introduces: (i) anatomy-aware and protocol-aware networks that personalize CT reconstruction to patient and scanner-specific features, (ii) multi-constraint projection losses that enforce consistency with CT measurements, and (iii) uncertainty-guided selective aggregation that weights clients by prediction confidence. Extensive experiments on the Mayo Clinic 2016 dataset demonstrate that ProFed achieves 42.56 dB PSNR with CNN backbones and 44.83 dB with Transformers, outperforming 11 federated learning baselines, including the physics-informed SCAN-PhysFed by +1.42 dB.
Task-Agnostic Continual Learning for Chest Radiograph Classification
Feb 17, 2026Clinical deployment of chest radiograph classifiers requires models that can be updated as new datasets become available without retraining on previously ob- served data or degrading validated performance. We study, for the first time, a task-incremental continual learning setting for chest radiograph classification, in which heterogeneous chest X-ray datasets arrive sequentially and task identifiers are unavailable at inference. We propose a continual adapter-based routing learning strategy for Chest X-rays (CARL-XRay) that maintains a fixed high-capacity backbone and incrementally allocates lightweight task-specific adapters and classifier heads. A latent task selector operates on task-adapted features and leverages both current and historical context preserved through compact prototypes and feature-level experience replay. This design supports stable task identification and adaptation across sequential updates while avoiding raw-image storage. Experiments on large-scale public chest radiograph datasets demonstrate robust performance retention and reliable task-aware inference under continual dataset ingestion. CARL-XRay outperforms joint training under task-unknown deployment, achieving higher routing accuracy (75.0\% vs.\ 62.5\%), while maintaining competitive diagnostic performance with AUROC of 0.74 in the oracle setting with ground-truth task identity and 0.75 under task-unknown inference, using significantly fewer trainable parameters. Finally, the proposed framework provides a practical alternative to joint training and repeated full retraining in continual clinical deployment.
Variational Bayesian Flow Network for Graph Generation
Jan 30, 2026Graph generation aims to sample discrete node and edge attributes while satisfying coupled structural constraints. Diffusion models for graphs often adopt largely factorized forward-noising, and many flow-matching methods start from factorized reference noise and coordinate-wise interpolation, so node-edge coupling is not encoded by the generative geometry and must be recovered implicitly by the core network, which can be brittle after discrete decoding. Bayesian Flow Networks (BFNs) evolve distribution parameters and naturally support discrete generation. But classical BFNs typically rely on factorized beliefs and independent channels, which limit geometric evidence fusion. We propose Variational Bayesian Flow Network (VBFN), which performs a variational lifting to a tractable joint Gaussian variational belief family governed by structured precisions. Each Bayesian update reduces to solving a symmetric positive definite linear system, enabling coupled node and edge updates within a single fusion step. We construct sample-agnostic sparse precisions from a representation-induced dependency graph, thereby avoiding label leakage while enforcing node-edge consistency. On synthetic and molecular graph datasets, VBFN improves fidelity and diversity, and surpasses baseline methods.
Modeling Endogenous Logic: Causal Neuro-Symbolic Reasoning Model for Explainable Multi-Behavior Recommendation
Jan 29, 2026Existing multi-behavior recommendations tend to prioritize performance at the expense of explainability, while current explainable methods suffer from limited generalizability due to their reliance on external information. Neuro-Symbolic integration offers a promising avenue for explainability by combining neural networks with symbolic logic rule reasoning. Concurrently, we posit that user behavior chains inherently embody an endogenous logic suitable for explicit reasoning. However, these observational multiple behaviors are plagued by confounders, causing models to learn spurious correlations. By incorporating causal inference into this Neuro-Symbolic framework, we propose a novel Causal Neuro-Symbolic Reasoning model for Explainable Multi-Behavior Recommendation (CNRE). CNRE operationalizes the endogenous logic by simulating a human-like decision-making process. Specifically, CNRE first employs hierarchical preference propagation to capture heterogeneous cross-behavior dependencies. Subsequently, it models the endogenous logic rule implicit in the user's behavior chain based on preference strength, and adaptively dispatches to the corresponding neural-logic reasoning path (e.g., conjunction, disjunction). This process generates an explainable causal mediator that approximates an ideal state isolated from confounding effects. Extensive experiments on three large-scale datasets demonstrate CNRE's significant superiority over state-of-the-art baselines, offering multi-level explainability from model design and decision process to recommendation results.
Multimodal Visual Surrogate Compression for Alzheimer's Disease Classification
Jan 29, 2026High-dimensional structural MRI (sMRI) images are widely used for Alzheimer's Disease (AD) diagnosis. Most existing methods for sMRI representation learning rely on 3D architectures (e.g., 3D CNNs), slice-wise feature extraction with late aggregation, or apply training-free feature extractions using 2D foundation models (e.g., DINO). However, these three paradigms suffer from high computational cost, loss of cross-slice relations, and limited ability to extract discriminative features, respectively. To address these challenges, we propose Multimodal Visual Surrogate Compression (MVSC). It learns to compress and adapt large 3D sMRI volumes into compact 2D features, termed as visual surrogates, which are better aligned with frozen 2D foundation models to extract powerful representations for final AD classification. MVSC has two key components: a Volume Context Encoder that captures global cross-slice context under textual guidance, and an Adaptive Slice Fusion module that aggregates slice-level information in a text-enhanced, patch-wise manner. Extensive experiments on three large-scale Alzheimer's disease benchmarks demonstrate our MVSC performs favourably on both binary and multi-class classification tasks compared against state-of-the-art methods.
PCEvo: Path-Consistent Molecular Representation via Virtual Evolutionary
Jan 27, 2026Molecular representation learning aims to learn vector embeddings that capture molecular structure and geometry, thereby enabling property prediction and downstream scientific applications. In many AI for science tasks, labeled data are expensive to obtain and therefore limited in availability. Under the few-shot setting, models trained with scarce supervision often learn brittle structure-property relationships, resulting in substantially higher prediction errors and reduced generalization to unseen molecules. To address this limitation, we propose PCEvo, a path-consistent representation method that learns from virtual paths through dynamic structural evolution. PCEvo enumerates multiple chemically feasible edit paths between retrieved similar molecular pairs under topological dependency constraints. It transforms the labels of the two molecules into stepwise supervision along each virtual evolutionary path. It introduces a path-consistency objective that enforces prediction invariance across alternative paths connecting the same two molecules. Comprehensive experiments on the QM9 and MoleculeNet datasets demonstrate that PCEvo substantially improves the few-shot generalization performance of baseline methods. The code is available at https://anonymous.4open.science/r/PCEvo-4BF2.
PMPBench: A Paired Multi-Modal Pan-Cancer Benchmark for Medical Image Synthesis
Jan 22, 2026Contrast medium plays a pivotal role in radiological imaging, as it amplifies lesion conspicuity and improves detection for the diagnosis of tumor-related diseases. However, depending on the patient's health condition or the medical resources available, the use of contrast medium is not always feasible. Recent work has explored AI-based image translation to synthesize contrast-enhanced images directly from non-contrast scans, aims to reduce side effects and streamlines clinical workflows. Progress in this direction has been constrained by data limitations: (1) existing public datasets focus almost exclusively on brain-related paired MR modalities; (2) other collections include partially paired data but suffer from missing modalities/timestamps and imperfect spatial alignment; (3) explicit labeling of CT vs. CTC or DCE phases is often absent; (4) substantial resources remain private. To bridge this gap, we introduce the first public, fully paired, pan-cancer medical imaging dataset spanning 11 human organs. The MR data include complete dynamic contrast-enhanced (DCE) sequences covering all three phases (DCE1-DCE3), while the CT data provide paired non-contrast and contrast-enhanced acquisitions (CTC). The dataset is curated for anatomical correspondence, enabling rigorous evaluation of 1-to-1, N-to-1, and N-to-N translation settings (e.g., predicting DCE phases from non-contrast inputs). Built upon this resource, we establish a comprehensive benchmark. We report results from representative baselines of contemporary image-to-image translation. We release the dataset and benchmark to catalyze research on safe, effective contrast synthesis, with direct relevance to multi-organ oncology imaging workflows. Our code and dataset are publicly available at https://github.com/YifanChen02/PMPBench.
Foundation Models in Biomedical Imaging: Turning Hype into Reality
Dec 17, 2025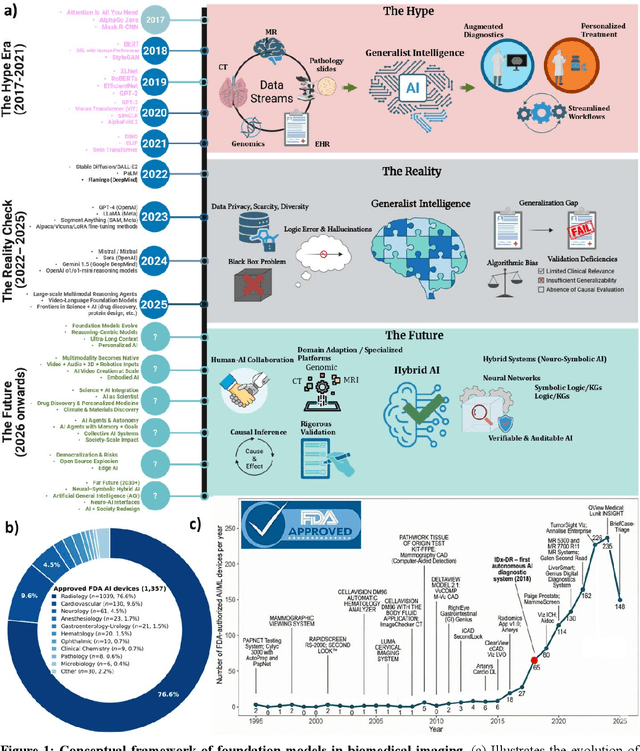
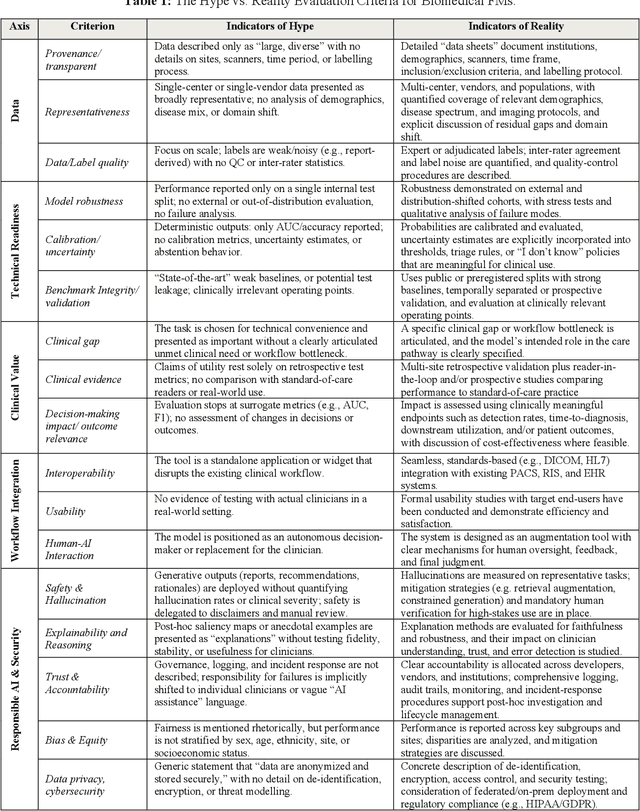
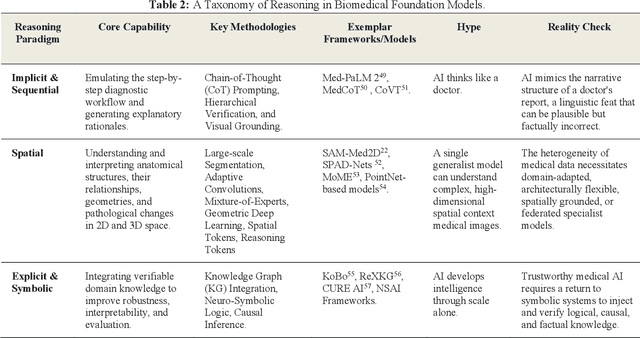
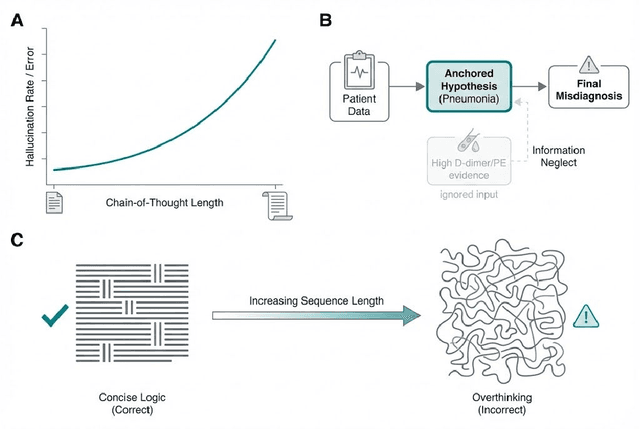
Foundation models (FMs) are driving a prominent shift in artificial intelligence across different domains, including biomedical imaging. These models are designed to move beyond narrow pattern recognition towards emulating sophisticated clinical reasoning, understanding complex spatial relationships, and integrating multimodal data with unprecedented flexibility. However, a critical gap exists between this potential and the current reality, where the clinical evaluation and deployment of FMs are hampered by significant challenges. Herein, we critically assess the current state-of-the-art, analyzing hype by examining the core capabilities and limitations of FMs in the biomedical domain. We also provide a taxonomy of reasoning, ranging from emulated sequential logic and spatial understanding to the integration of explicit symbolic knowledge, to evaluate whether these models exhibit genuine cognition or merely mimic surface-level patterns. We argue that a critical frontier lies beyond statistical correlation, in the pursuit of causal inference, which is essential for building robust models that understand cause and effect. Furthermore, we discuss the paramount issues in deployment stemming from trustworthiness, bias, and safety, dissecting the challenges of algorithmic bias, data bias and privacy, and model hallucinations. We also draw attention to the need for more inclusive, rigorous, and clinically relevant validation frameworks to ensure their safe and ethical application. We conclude that while the vision of autonomous AI-doctors remains distant, the immediate reality is the emergence of powerful technology and assistive tools that would benefit clinical practice. The future of FMs in biomedical imaging hinges not on scale alone, but on developing hybrid, causally aware, and verifiably safe systems that augment, rather than replace, human expertise.
Adversarial Attacks Against Automated Fact-Checking: A Survey
Sep 10, 2025In an era where misinformation spreads freely, fact-checking (FC) plays a crucial role in verifying claims and promoting reliable information. While automated fact-checking (AFC) has advanced significantly, existing systems remain vulnerable to adversarial attacks that manipulate or generate claims, evidence, or claim-evidence pairs. These attacks can distort the truth, mislead decision-makers, and ultimately undermine the reliability of FC models. Despite growing research interest in adversarial attacks against AFC systems, a comprehensive, holistic overview of key challenges remains lacking. These challenges include understanding attack strategies, assessing the resilience of current models, and identifying ways to enhance robustness. This survey provides the first in-depth review of adversarial attacks targeting FC, categorizing existing attack methodologies and evaluating their impact on AFC systems. Additionally, we examine recent advancements in adversary-aware defenses and highlight open research questions that require further exploration. Our findings underscore the urgent need for resilient FC frameworks capable of withstanding adversarial manipulations in pursuit of preserving high verification accuracy.