 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeContextLens: Modeling Imperfect Privacy and Safety Context for Legal Compliance
Apr 14, 2026Individuals' concerns about data privacy and AI safety are highly contextualized and extend beyond sensitive patterns. Addressing these issues requires reasoning about the context to identify and mitigate potential risks. Though researchers have widely explored using large language models (LLMs) as evaluators for contextualized safety and privacy assessments, these efforts typically assume the availability of complete and clear context, whereas real-world contexts tend to be ambiguous and incomplete. In this paper, we propose ContextLens, a semi-rule-based framework that leverages LLMs to ground the input context in the legal domain and explicitly identify both known and unknown factors for legal compliance. Instead of directly assessing safety outcomes, our ContextLens instructs LLMs to answer a set of crafted questions that span over applicability, general principles and detailed provisions to assess compliance with pre-defined priorities and rules. We conduct extensive experiments on existing compliance benchmarks that cover the General Data Protection Regulation (GDPR) and the EU AI Act. The results suggest that our ContextLens can significantly improve LLMs' compliance assessment and surpass existing baselines without any training. Additionally, our ContextLens can further identify the ambiguous and missing factors.
OmniCompliance-100K: A Multi-Domain, Rule-Grounded, Real-World Safety Compliance Dataset
Mar 14, 2026Ensuring the safety and compliance of large language models (LLMs) is of paramount importance. However, existing LLM safety datasets often rely on ad-hoc taxonomies for data generation and suffer from a significant shortage of rule-grounded, real-world cases that are essential for robustly protecting LLMs. In this work, we address this critical gap by constructing a comprehensive safety dataset from a compliance perspective. Using a powerful web-searching agent, we collect a rule-grounded, real-world case dataset OmniCompliance-100K, sourced from multi-domain authoritative references. The dataset spans 74 regulations and policies across a wide range of domains, including security and privacy regulations, content safety and user data privacy policies from leading AI companies and social media platforms, financial security requirements, medical device risk management standards, educational integrity guidelines, and protections of fundamental human rights. In total, our dataset contains 12,985 distinct rules and 106,009 associated real-world compliance cases. Our analysis confirms a strong alignment between the rules and their corresponding cases. We further conduct extensive benchmarking experiments to evaluate the safety and compliance capabilities of advanced LLMs across different model scales. Our experiments reveal several interesting findings that have great potential to offer valuable insights for future LLM safety research.
Variational Bayesian Flow Network for Graph Generation
Jan 30, 2026Graph generation aims to sample discrete node and edge attributes while satisfying coupled structural constraints. Diffusion models for graphs often adopt largely factorized forward-noising, and many flow-matching methods start from factorized reference noise and coordinate-wise interpolation, so node-edge coupling is not encoded by the generative geometry and must be recovered implicitly by the core network, which can be brittle after discrete decoding. Bayesian Flow Networks (BFNs) evolve distribution parameters and naturally support discrete generation. But classical BFNs typically rely on factorized beliefs and independent channels, which limit geometric evidence fusion. We propose Variational Bayesian Flow Network (VBFN), which performs a variational lifting to a tractable joint Gaussian variational belief family governed by structured precisions. Each Bayesian update reduces to solving a symmetric positive definite linear system, enabling coupled node and edge updates within a single fusion step. We construct sample-agnostic sparse precisions from a representation-induced dependency graph, thereby avoiding label leakage while enforcing node-edge consistency. On synthetic and molecular graph datasets, VBFN improves fidelity and diversity, and surpasses baseline methods.
PCEvo: Path-Consistent Molecular Representation via Virtual Evolutionary
Jan 27, 2026Molecular representation learning aims to learn vector embeddings that capture molecular structure and geometry, thereby enabling property prediction and downstream scientific applications. In many AI for science tasks, labeled data are expensive to obtain and therefore limited in availability. Under the few-shot setting, models trained with scarce supervision often learn brittle structure-property relationships, resulting in substantially higher prediction errors and reduced generalization to unseen molecules. To address this limitation, we propose PCEvo, a path-consistent representation method that learns from virtual paths through dynamic structural evolution. PCEvo enumerates multiple chemically feasible edit paths between retrieved similar molecular pairs under topological dependency constraints. It transforms the labels of the two molecules into stepwise supervision along each virtual evolutionary path. It introduces a path-consistency objective that enforces prediction invariance across alternative paths connecting the same two molecules. Comprehensive experiments on the QM9 and MoleculeNet datasets demonstrate that PCEvo substantially improves the few-shot generalization performance of baseline methods. The code is available at https://anonymous.4open.science/r/PCEvo-4BF2.
Safety Compliance: Rethinking LLM Safety Reasoning through the Lens of Compliance
Sep 26, 2025The proliferation of Large Language Models (LLMs) has demonstrated remarkable capabilities, elevating the critical importance of LLM safety. However, existing safety methods rely on ad-hoc taxonomy and lack a rigorous, systematic protection, failing to ensure safety for the nuanced and complex behaviors of modern LLM systems. To address this problem, we solve LLM safety from legal compliance perspectives, named safety compliance. In this work, we posit relevant established legal frameworks as safety standards for defining and measuring safety compliance, including the EU AI Act and GDPR, which serve as core legal frameworks for AI safety and data security in Europe. To bridge the gap between LLM safety and legal compliance, we first develop a new benchmark for safety compliance by generating realistic LLM safety scenarios seeded with legal statutes. Subsequently, we align Qwen3-8B using Group Policy Optimization (GRPO) to construct a safety reasoner, Compliance Reasoner, which effectively aligns LLMs with legal standards to mitigate safety risks. Our comprehensive experiments demonstrate that the Compliance Reasoner achieves superior performance on the new benchmark, with average improvements of +10.45% for the EU AI Act and +11.85% for GDPR.
Zero-Shot Learning with Subsequence Reordering Pretraining for Compound-Protein Interaction
Jul 28, 2025Given the vastness of chemical space and the ongoing emergence of previously uncharacterized proteins, zero-shot compound-protein interaction (CPI) prediction better reflects the practical challenges and requirements of real-world drug development. Although existing methods perform adequately during certain CPI tasks, they still face the following challenges: (1) Representation learning from local or complete protein sequences often overlooks the complex interdependencies between subsequences, which are essential for predicting spatial structures and binding properties. (2) Dependence on large-scale or scarce multimodal protein datasets demands significant training data and computational resources, limiting scalability and efficiency. To address these challenges, we propose a novel approach that pretrains protein representations for CPI prediction tasks using subsequence reordering, explicitly capturing the dependencies between protein subsequences. Furthermore, we apply length-variable protein augmentation to ensure excellent pretraining performance on small training datasets. To evaluate the model's effectiveness and zero-shot learning ability, we combine it with various baseline methods. The results demonstrate that our approach can improve the baseline model's performance on the CPI task, especially in the challenging zero-shot scenario. Compared to existing pre-training models, our model demonstrates superior performance, particularly in data-scarce scenarios where training samples are limited. Our implementation is available at https://github.com/Hoch-Zhang/PSRP-CPI.
Antibody Design and Optimization with Multi-scale Equivariant Graph Diffusion Models for Accurate Complex Antigen Binding
Jun 26, 2025Antibody design remains a critical challenge in therapeutic and diagnostic development, particularly for complex antigens with diverse binding interfaces. Current computational methods face two main limitations: (1) capturing geometric features while preserving symmetries, and (2) generalizing novel antigen interfaces. Despite recent advancements, these methods often fail to accurately capture molecular interactions and maintain structural integrity. To address these challenges, we propose \textbf{AbMEGD}, an end-to-end framework integrating \textbf{M}ulti-scale \textbf{E}quivariant \textbf{G}raph \textbf{D}iffusion for antibody sequence and structure co-design. Leveraging advanced geometric deep learning, AbMEGD combines atomic-level geometric features with residue-level embeddings, capturing local atomic details and global sequence-structure interactions. Its E(3)-equivariant diffusion method ensures geometric precision, computational efficiency, and robust generalizability for complex antigens. Furthermore, experiments using the SAbDab database demonstrate a 10.13\% increase in amino acid recovery, 3.32\% rise in improvement percentage, and a 0.062~\AA\ reduction in root mean square deviation within the critical CDR-H3 region compared to DiffAb, a leading antibody design model. These results highlight AbMEGD's ability to balance structural integrity with improved functionality, establishing a new benchmark for sequence-structure co-design and affinity optimization. The code is available at: https://github.com/Patrick221215/AbMEGD.
MCIP: Protecting MCP Safety via Model Contextual Integrity Protocol
May 21, 2025As Model Context Protocol (MCP) introduces an easy-to-use ecosystem for users and developers, it also brings underexplored safety risks. Its decentralized architecture, which separates clients and servers, poses unique challenges for systematic safety analysis. This paper proposes a novel framework to enhance MCP safety. Guided by the MAESTRO framework, we first analyze the missing safety mechanisms in MCP, and based on this analysis, we propose the Model Contextual Integrity Protocol (MCIP), a refined version of MCP that addresses these gaps. Next, we develop a fine-grained taxonomy that captures a diverse range of unsafe behaviors observed in MCP scenarios. Building on this taxonomy, we develop benchmark and training data that support the evaluation and improvement of LLMs' capabilities in identifying safety risks within MCP interactions. Leveraging the proposed benchmark and training data, we conduct extensive experiments on state-of-the-art LLMs. The results highlight LLMs' vulnerabilities in MCP interactions and demonstrate that our approach substantially improves their safety performance.
Context Reasoner: Incentivizing Reasoning Capability for Contextualized Privacy and Safety Compliance via Reinforcement Learning
May 20, 2025While Large Language Models (LLMs) exhibit remarkable capabilities, they also introduce significant safety and privacy risks. Current mitigation strategies often fail to preserve contextual reasoning capabilities in risky scenarios. Instead, they rely heavily on sensitive pattern matching to protect LLMs, which limits the scope. Furthermore, they overlook established safety and privacy standards, leading to systemic risks for legal compliance. To address these gaps, we formulate safety and privacy issues into contextualized compliance problems following the Contextual Integrity (CI) theory. Under the CI framework, we align our model with three critical regulatory standards: GDPR, EU AI Act, and HIPAA. Specifically, we employ reinforcement learning (RL) with a rule-based reward to incentivize contextual reasoning capabilities while enhancing compliance with safety and privacy norms. Through extensive experiments, we demonstrate that our method not only significantly enhances legal compliance (achieving a +17.64% accuracy improvement in safety/privacy benchmarks) but also further improves general reasoning capability. For OpenThinker-7B, a strong reasoning model that significantly outperforms its base model Qwen2.5-7B-Instruct across diverse subjects, our method enhances its general reasoning capabilities, with +2.05% and +8.98% accuracy improvement on the MMLU and LegalBench benchmark, respectively.
DrugPilot: LLM-based Parameterized Reasoning Agent for Drug Discovery
May 20, 2025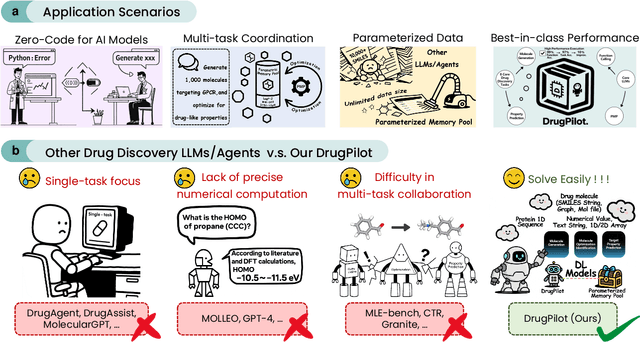


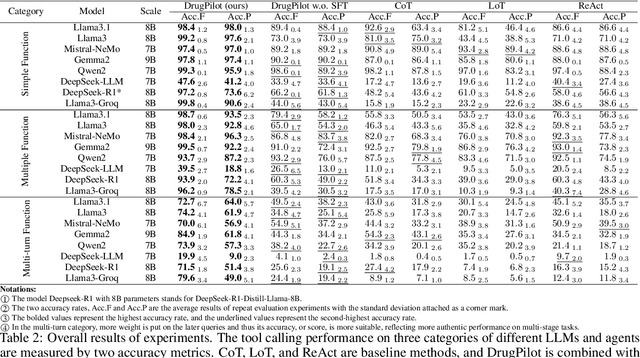
In the field of AI4Science, large-scale language models (LLMs) show great potential to parse complex scientific semantics, integrate cross-disciplinary knowledge, and assist critical task research. However, in the field of drug discovery, despite the optimization through professional data pre-training, context window expansion, and internet search, the existing LLMs are still facing challenges such as massive multi-modal and heterogeneous data processing, domain knowledge dynamic updating delay, and insufficient confidence in predicting the results of complex computational tasks. To address these challenges, we propose the DrugPilot, an LLM-based agent with parameterized reasoning for drug discovery. DrugPilot addresses key limitations of traditional end-to-end LLM prediction approaches through its parametric inference architecture. This agent system supports major phases of the drug discovery pipeline, facilitating automated planning and execution of multi-stage research tasks. To address the critical challenge of multi-modal drug data analysis (incorporating both public datasets and user-submitted data), we developed an interactive parameterized memory pool. This innovative component standardizes real-world drug data into parametric representations, simultaneously enabling efficient knowledge retrieval in multi-turn dialogue while mitigating the information loss inherent in text-based data transmission. Additionally, we created a drug instruct dataset across 8 essential drug discovery tasks for model fine-tuning and evaluation. Based on the Berkeley function calling evaluation framework, DrugPilot demonstrated the most advanced tool calling capabilities on our drug discovery tool instruction dataset, outperforming existing agents (e.g., ReAct, LoT). Specifically, it achieves task completion rates of 98.0%, 93.5%, and 64.0% on simple, multiple, and multi-turn tasks, respectively.