 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe First Challenge on Remote Sensing Infrared Image Super-Resolution at NTIRE 2026: Benchmark Results and Method Overview
Apr 23, 2026This paper presents the NTIRE 2026 Remote Sensing Infrared Image Super-Resolution (x4) Challenge, one of the associated challenges of NTIRE 2026. The challenge aims to recover high-resolution (HR) infrared images from low-resolution (LR) inputs generated through bicubic downsampling with a x4 scaling factor. The objective is to develop effective models or solutions that achieve state-of-the-art performance for infrared image SR in remote sensing scenarios. To reflect the characteristics of infrared data and practical application needs, the challenge adopts a single-track setting. A total of 115 participants registered for the competition, with 13 teams submitting valid entries. This report summarizes the challenge design, dataset, evaluation protocol, main results, and the representative methods of each team. The challenge serves as a benchmark to advance research in infrared image super-resolution and promote the development of effective solutions for real-world remote sensing applications.
Dual-Branch Remote Sensing Infrared Image Super-Resolution
Apr 11, 2026Remote sensing infrared image super-resolution aims to recover sharper thermal observations from low-resolution inputs while preserving target contours, scene layout, and radiometric stability. Unlike visible-image super-resolution, thermal imagery is weakly textured and more sensitive to unstable local sharpening, which makes complementary local and global modeling especially important. This paper presents our solution to the NTIRE 2026 Infrared Image Super-Resolution Challenge, a dual-branch system that combines a HAT-L branch and a MambaIRv2-L branch. The inference pipeline applies test-time local conversion on HAT, eight-way self-ensemble on MambaIRv2, and fixed equal-weight image-space fusion. We report both the official challenge score and a reproducible evaluation on 12 synthetic times-four thermal samples derived from Caltech Aerial RGB-Thermal, on which the fused output outperforms either single branch in PSNR, SSIM, and the overall Score. The results suggest that infrared super-resolution benefits from explicit complementarity between locally strong transformer restoration and globally stable state-space modeling.
The Tenth NTIRE 2025 Image Denoising Challenge Report
Apr 16, 2025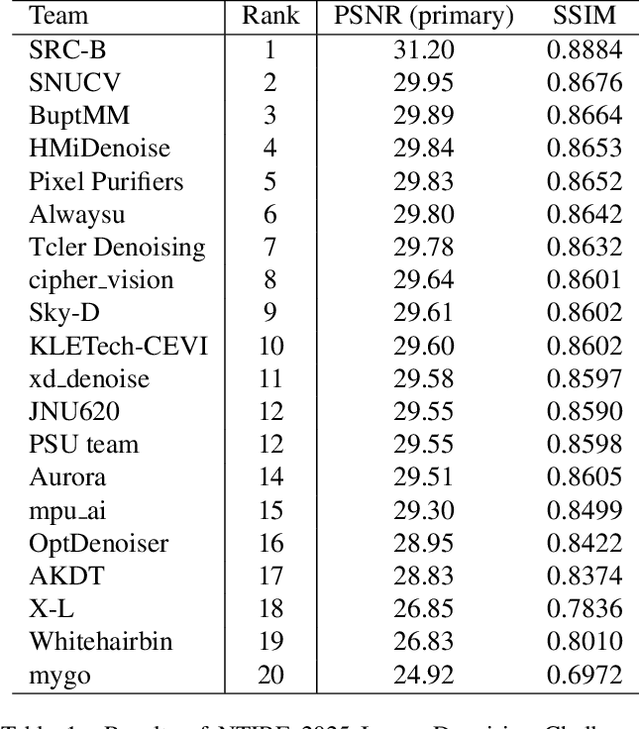

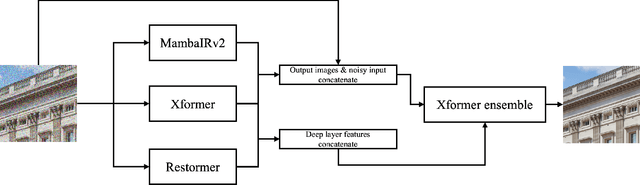

This paper presents an overview of the NTIRE 2025 Image Denoising Challenge ({\sigma} = 50), highlighting the proposed methodologies and corresponding results. The primary objective is to develop a network architecture capable of achieving high-quality denoising performance, quantitatively evaluated using PSNR, without constraints on computational complexity or model size. The task assumes independent additive white Gaussian noise (AWGN) with a fixed noise level of 50. A total of 290 participants registered for the challenge, with 20 teams successfully submitting valid results, providing insights into the current state-of-the-art in image denoising.
The Tenth NTIRE 2025 Efficient Super-Resolution Challenge Report
Apr 14, 2025This paper presents a comprehensive review of the NTIRE 2025 Challenge on Single-Image Efficient Super-Resolution (ESR). The challenge aimed to advance the development of deep models that optimize key computational metrics, i.e., runtime, parameters, and FLOPs, while achieving a PSNR of at least 26.90 dB on the $\operatorname{DIV2K\_LSDIR\_valid}$ dataset and 26.99 dB on the $\operatorname{DIV2K\_LSDIR\_test}$ dataset. A robust participation saw \textbf{244} registered entrants, with \textbf{43} teams submitting valid entries. This report meticulously analyzes these methods and results, emphasizing groundbreaking advancements in state-of-the-art single-image ESR techniques. The analysis highlights innovative approaches and establishes benchmarks for future research in the field.
Purrfessor: A Fine-tuned Multimodal LLaVA Diet Health Chatbot
Nov 22, 2024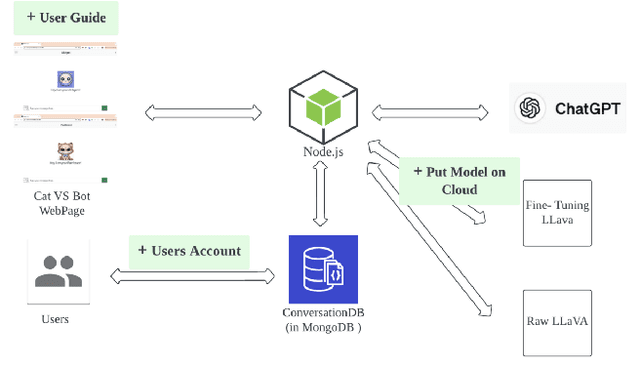
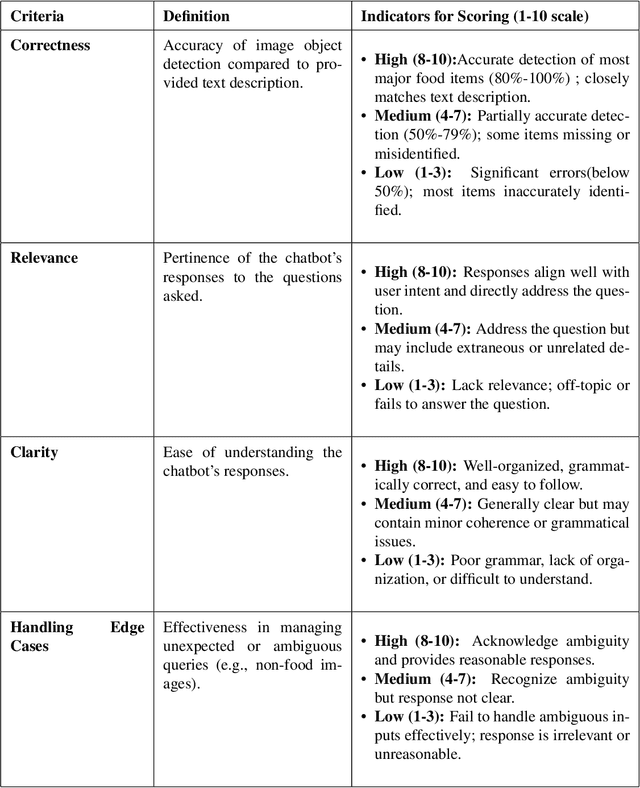
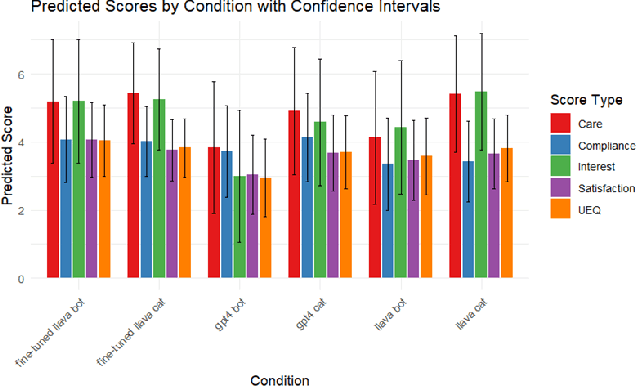
This study introduces Purrfessor, an innovative AI chatbot designed to provide personalized dietary guidance through interactive, multimodal engagement. Leveraging the Large Language-and-Vision Assistant (LLaVA) model fine-tuned with food and nutrition data and a human-in-the-loop approach, Purrfessor integrates visual meal analysis with contextual advice to enhance user experience and engagement. We conducted two studies to evaluate the chatbot's performance and user experience: (a) simulation assessments and human validation were conducted to examine the performance of the fine-tuned model; (b) a 2 (Profile: Bot vs. Pet) by 3 (Model: GPT-4 vs. LLaVA vs. Fine-tuned LLaVA) experiment revealed that Purrfessor significantly enhanced users' perceptions of care ($\beta = 1.59$, $p = 0.04$) and interest ($\beta = 2.26$, $p = 0.01$) compared to the GPT-4 bot. Additionally, user interviews highlighted the importance of interaction design details, emphasizing the need for responsiveness, personalization, and guidance to improve user engagement.
Chemical Language Model Linker: blending text and molecules with modular adapters
Oct 26, 2024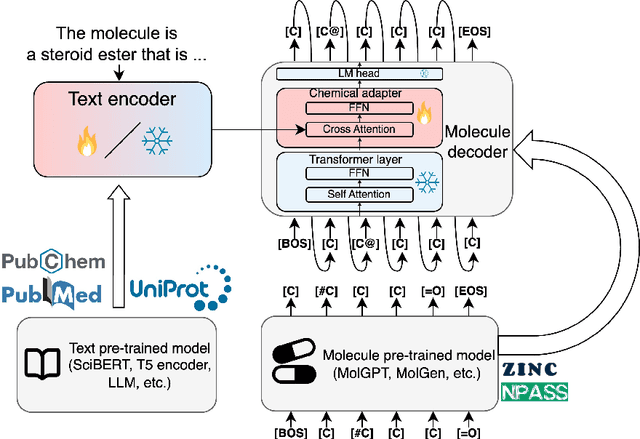
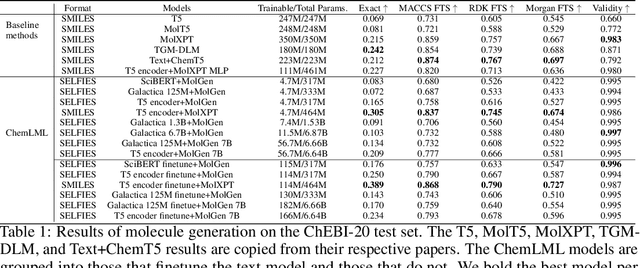
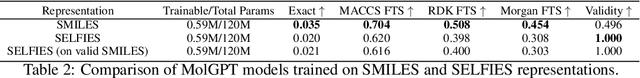
The development of large language models and multi-modal models has enabled the appealing idea of generating novel molecules from text descriptions. Generative modeling would shift the paradigm from relying on large-scale chemical screening to find molecules with desired properties to directly generating those molecules. However, multi-modal models combining text and molecules are often trained from scratch, without leveraging existing high-quality pretrained models. That approach consumes more computational resources and prohibits model scaling. In contrast, we propose a lightweight adapter-based strategy named Chemical Language Model Linker (ChemLML). ChemLML blends the two single domain models and obtains conditional molecular generation from text descriptions while still operating in the specialized embedding spaces of the molecular domain. ChemLML can tailor diverse pretrained text models for molecule generation by training relatively few adapter parameters. We find that the choice of molecular representation used within ChemLML, SMILES versus SELFIES, has a strong influence on conditional molecular generation performance. SMILES is often preferable despite not guaranteeing valid molecules. We raise issues in using the large PubChem dataset of molecules and their associated descriptions for evaluating molecule generation and provide a filtered version of the dataset as a generation test set. To demonstrate how ChemLML could be used in practice, we generate candidate protein inhibitors and use docking to assess their quality.
PepHarmony: A Multi-View Contrastive Learning Framework for Integrated Sequence and Structure-Based Peptide Encoding
Jan 21, 2024



Recent advances in protein language models have catalyzed significant progress in peptide sequence representation. Despite extensive exploration in this field, pre-trained models tailored for peptide-specific needs remain largely unaddressed due to the difficulty in capturing the complex and sometimes unstable structures of peptides. This study introduces a novel multi-view contrastive learning framework PepHarmony for the sequence-based peptide encoding task. PepHarmony innovatively combines both sequence- and structure-level information into a sequence-level encoding module through contrastive learning. We carefully select datasets from the Protein Data Bank (PDB) and AlphaFold database to encompass a broad spectrum of peptide sequences and structures. The experimental data highlights PepHarmony's exceptional capability in capturing the intricate relationship between peptide sequences and structures compared with the baseline and fine-tuned models. The robustness of our model is confirmed through extensive ablation studies, which emphasize the crucial roles of contrastive loss and strategic data sorting in enhancing predictive performance. The proposed PepHarmony framework serves as a notable contribution to peptide representations, and offers valuable insights for future applications in peptide drug discovery and peptide engineering. We have made all the source code utilized in this study publicly accessible via GitHub at https://github.com/zhangruochi/PepHarmony or http://www.healthinformaticslab.org/supp/.
Structure-informed Language Models Are Protein Designers
Feb 09, 2023This paper demonstrates that language models are strong structure-based protein designers. We present LM-Design, a generic approach to reprogramming sequence-based protein language models (pLMs), that have learned massive sequential evolutionary knowledge from the universe of natural protein sequences, to acquire an immediate capability to design preferable protein sequences for given folds. We conduct a structural surgery on pLMs, where a lightweight structural adapter is implanted into pLMs and endows it with structural awareness. During inference, iterative refinement is performed to effectively optimize the generated protein sequences. Experiments show that LM-Design improves the state-of-the-art results by a large margin, leading to up to 4% to 12% accuracy gains in sequence recovery (e.g., 55.65%/56.63% on CATH 4.2/4.3 single-chain benchmarks, and >60% when designing protein complexes). We provide extensive and in-depth analyses, which verify that LM-Design can (1) indeed leverage both structural and sequential knowledge to accurately handle structurally non-deterministic regions, (2) benefit from scaling data and model size, and (3) generalize to other proteins (e.g., antibodies and de novo proteins)
Improved Drug-target Interaction Prediction with Intermolecular Graph Transformer
Oct 15, 2021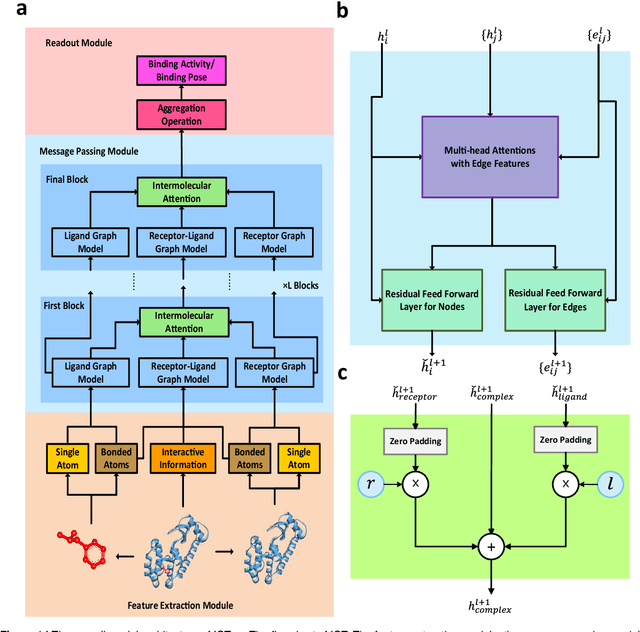
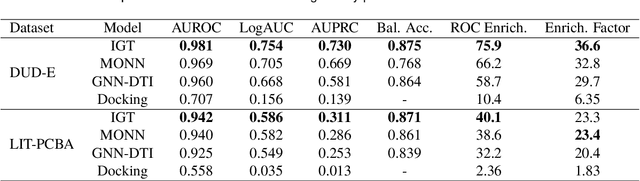
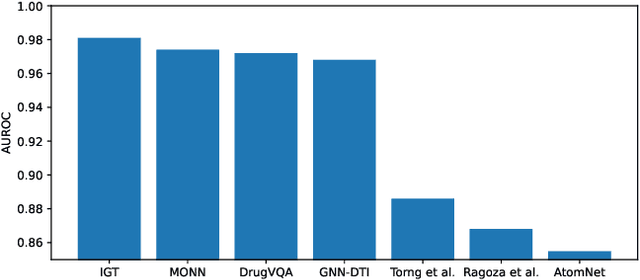

The identification of active binding drugs for target proteins (termed as drug-target interaction prediction) is the key challenge in virtual screening, which plays an essential role in drug discovery. Although recent deep learning-based approaches achieved better performance than molecular docking, existing models often neglect certain aspects of the intermolecular information, hindering the performance of prediction. We recognize this problem and propose a novel approach named Intermolecular Graph Transformer (IGT) that employs a dedicated attention mechanism to model intermolecular information with a three-way Transformer-based architecture. IGT outperforms state-of-the-art approaches by 9.1% and 20.5% over the second best for binding activity and binding pose prediction respectively, and shows superior generalization ability to unseen receptor proteins. Furthermore, IGT exhibits promising drug screening ability against SARS-CoV-2 by identifying 83.1% active drugs that have been validated by wet-lab experiments with near-native predicted binding poses.