 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExploring zero-shot structure-based protein fitness prediction
Apr 23, 2025The ability to make zero-shot predictions about the fitness consequences of protein sequence changes with pre-trained machine learning models enables many practical applications. Such models can be applied for downstream tasks like genetic variant interpretation and protein engineering without additional labeled data. The advent of capable protein structure prediction tools has led to the availability of orders of magnitude more precomputed predicted structures, giving rise to powerful structure-based fitness prediction models. Through our experiments, we assess several modeling choices for structure-based models and their effects on downstream fitness prediction. Zero-shot fitness prediction models can struggle to assess the fitness landscape within disordered regions of proteins, those that lack a fixed 3D structure. We confirm the importance of matching protein structures to fitness assays and find that predicted structures for disordered regions can be misleading and affect predictive performance. Lastly, we evaluate an additional structure-based model on the ProteinGym substitution benchmark and show that simple multi-modal ensembles are strong baselines.
* 26 pages, 7 figures
Chemical Language Model Linker: blending text and molecules with modular adapters
Oct 26, 2024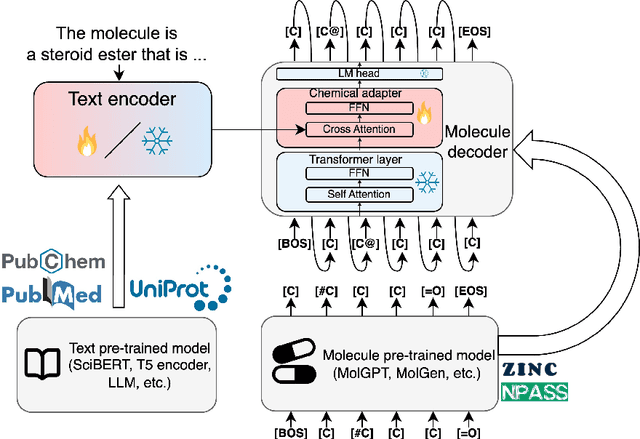
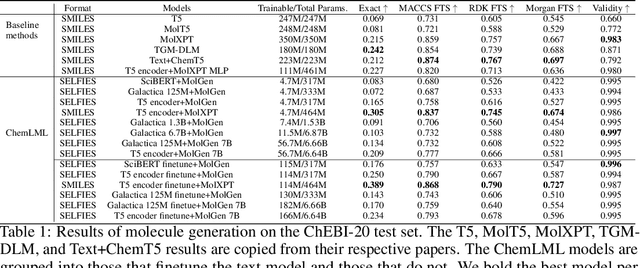
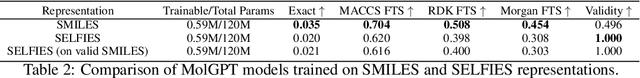
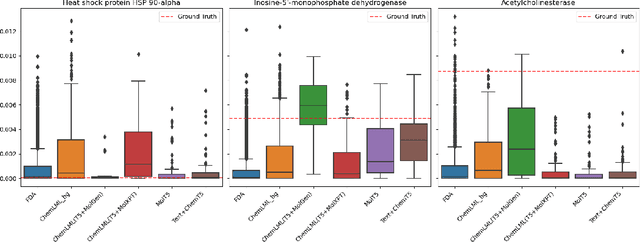
The development of large language models and multi-modal models has enabled the appealing idea of generating novel molecules from text descriptions. Generative modeling would shift the paradigm from relying on large-scale chemical screening to find molecules with desired properties to directly generating those molecules. However, multi-modal models combining text and molecules are often trained from scratch, without leveraging existing high-quality pretrained models. That approach consumes more computational resources and prohibits model scaling. In contrast, we propose a lightweight adapter-based strategy named Chemical Language Model Linker (ChemLML). ChemLML blends the two single domain models and obtains conditional molecular generation from text descriptions while still operating in the specialized embedding spaces of the molecular domain. ChemLML can tailor diverse pretrained text models for molecule generation by training relatively few adapter parameters. We find that the choice of molecular representation used within ChemLML, SMILES versus SELFIES, has a strong influence on conditional molecular generation performance. SMILES is often preferable despite not guaranteeing valid molecules. We raise issues in using the large PubChem dataset of molecules and their associated descriptions for evaluating molecule generation and provide a filtered version of the dataset as a generation test set. To demonstrate how ChemLML could be used in practice, we generate candidate protein inhibitors and use docking to assess their quality.
Product Manifold Representations for Learning on Biological Pathways
Jan 27, 2024



Machine learning models that embed graphs in non-Euclidean spaces have shown substantial benefits in a variety of contexts, but their application has not been studied extensively in the biological domain, particularly with respect to biological pathway graphs. Such graphs exhibit a variety of complex network structures, presenting challenges to existing embedding approaches. Learning high-quality embeddings for biological pathway graphs is important for researchers looking to understand the underpinnings of disease and train high-quality predictive models on these networks. In this work, we investigate the effects of embedding pathway graphs in non-Euclidean mixed-curvature spaces and compare against traditional Euclidean graph representation learning models. We then train a supervised model using the learned node embeddings to predict missing protein-protein interactions in pathway graphs. We find large reductions in distortion and boosts on in-distribution edge prediction performance as a result of using mixed-curvature embeddings and their corresponding graph neural network models. However, we find that mixed-curvature representations underperform existing baselines on out-of-distribution edge prediction performance suggesting that these representations may overfit to the training graph topology. We provide our mixed-curvature product GCN code at https://github.com/mcneela/Mixed-Curvature-GCN and our pathway analysis code at https://github.com/mcneela/Mixed-Curvature-Pathways.
A Text-guided Protein Design Framework
Feb 09, 2023

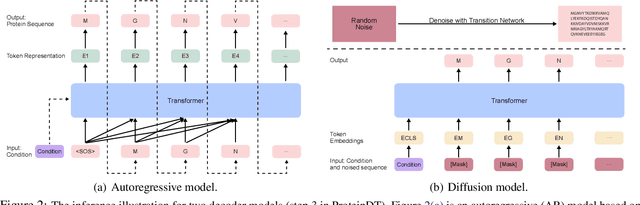

Current AI-assisted protein design mainly utilizes protein sequential and structural information. Meanwhile, there exists tremendous knowledge curated by humans in the text format describing proteins' high-level properties. Yet, whether the incorporation of such text data can help protein design tasks has not been explored. To bridge this gap, we propose ProteinDT, a multi-modal framework that leverages textual descriptions for protein design. ProteinDT consists of three subsequent steps: ProteinCLAP that aligns the representation of two modalities, a facilitator that generates the protein representation from the text modality, and a decoder that generates the protein sequences from the representation. To train ProteinDT, we construct a large dataset, SwissProtCLAP, with 441K text and protein pairs. We empirically verify the effectiveness of ProteinDT from three aspects: (1) consistently superior performance on four out of six protein property prediction benchmarks; (2) over 90% accuracy for text-guided protein generation; and (3) promising results for zero-shot text-guided protein editing.
Graph algorithms for predicting subcellular localization at the pathway level
Dec 12, 2022
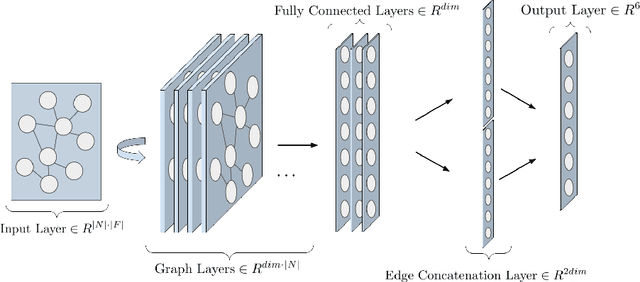
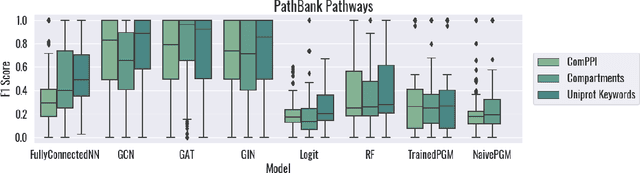
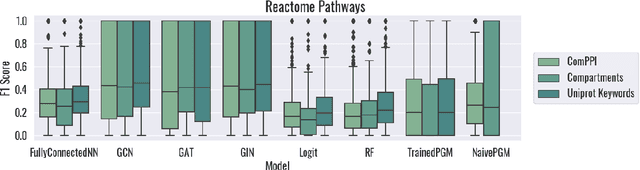
Protein subcellular localization is an important factor in normal cellular processes and disease. While many protein localization resources treat it as static, protein localization is dynamic and heavily influenced by biological context. Biological pathways are graphs that represent a specific biological context and can be inferred from large-scale data. We develop graph algorithms to predict the localization of all interactions in a biological pathway as an edge-labeling task. We compare a variety of models including graph neural networks, probabilistic graphical models, and discriminative classifiers for predicting localization annotations from curated pathway databases. We also perform a case study where we construct biological pathways and predict localizations of human fibroblasts undergoing viral infection. Pathway localization prediction is a promising approach for integrating publicly available localization data into the analysis of large-scale biological data.
* 35 pages, 14 figures
Ten Quick Tips for Deep Learning in Biology
May 29, 2021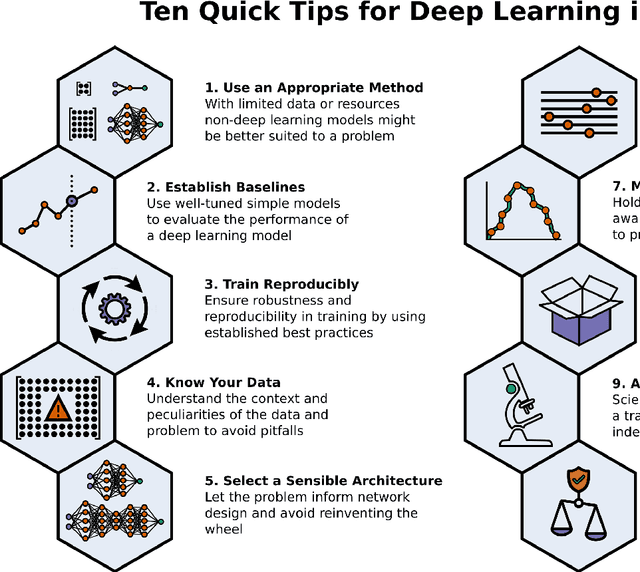
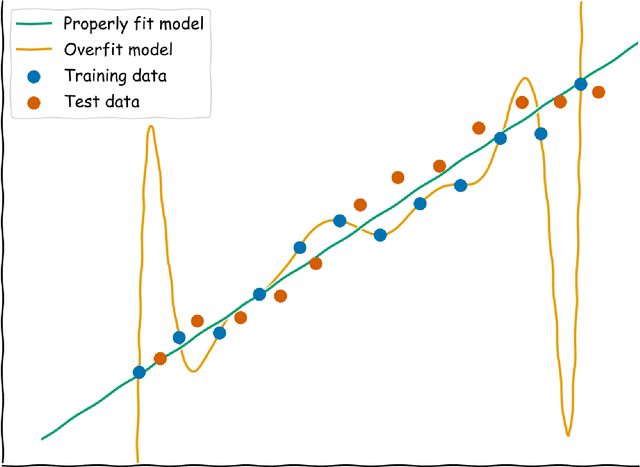
Machine learning is a modern approach to problem-solving and task automation. In particular, machine learning is concerned with the development and applications of algorithms that can recognize patterns in data and use them for predictive modeling. Artificial neural networks are a particular class of machine learning algorithms and models that evolved into what is now described as deep learning. Given the computational advances made in the last decade, deep learning can now be applied to massive data sets and in innumerable contexts. Therefore, deep learning has become its own subfield of machine learning. In the context of biological research, it has been increasingly used to derive novel insights from high-dimensional biological data. To make the biological applications of deep learning more accessible to scientists who have some experience with machine learning, we solicited input from a community of researchers with varied biological and deep learning interests. These individuals collaboratively contributed to this manuscript's writing using the GitHub version control platform and the Manubot manuscript generation toolset. The goal was to articulate a practical, accessible, and concise set of guidelines and suggestions to follow when using deep learning. In the course of our discussions, several themes became clear: the importance of understanding and applying machine learning fundamentals as a baseline for utilizing deep learning, the necessity for extensive model comparisons with careful evaluation, and the need for critical thought in interpreting results generated by deep learning, among others.
Inferring Signaling Pathways with Probabilistic Programming
May 28, 2020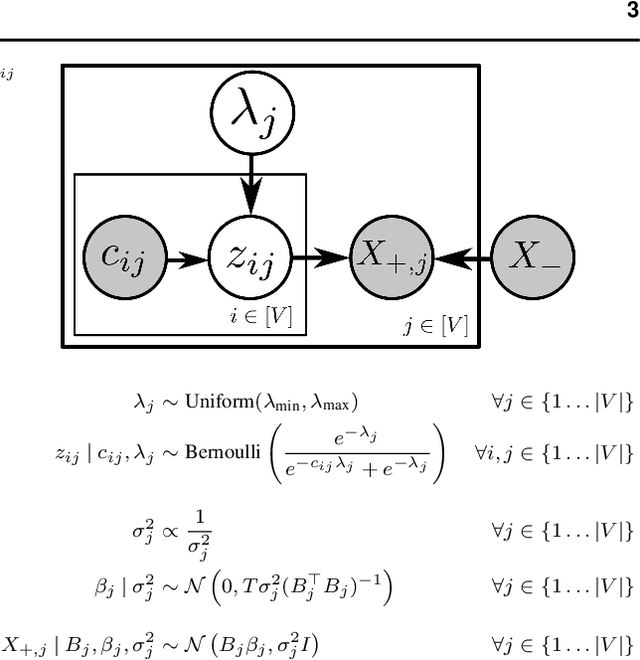
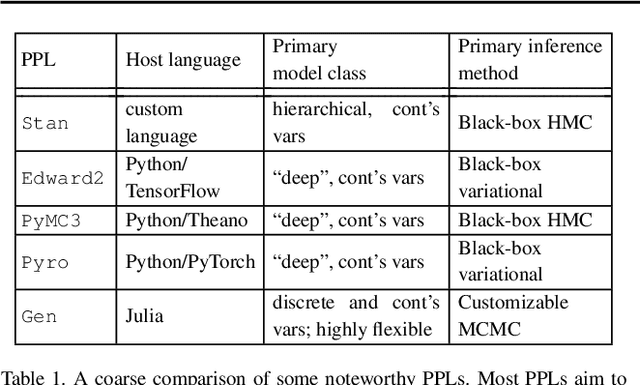
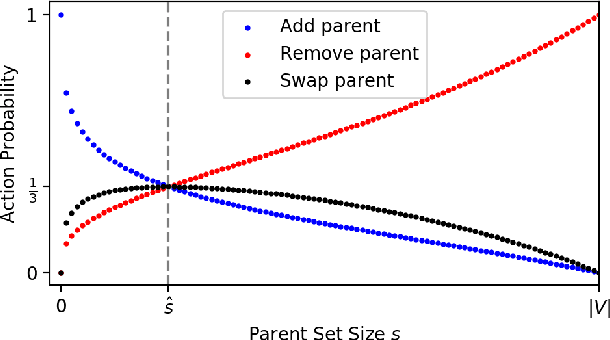
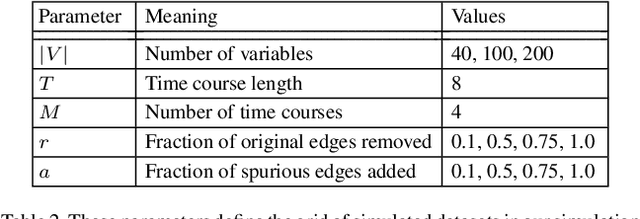
Cells regulate themselves via dizzyingly complex biochemical processes called signaling pathways. These are usually depicted as a network, where nodes represent proteins and edges indicate their influence on each other. In order to understand diseases and therapies at the cellular level, it is crucial to have an accurate understanding of the signaling pathways at work. Since signaling pathways can be modified by disease, the ability to infer signaling pathways from condition- or patient-specific data is highly valuable. A variety of techniques exist for inferring signaling pathways. We build on past works that formulate signaling pathway inference as a Dynamic Bayesian Network structure estimation problem on phosphoproteomic time course data. We take a Bayesian approach, using Markov Chain Monte Carlo to estimate a posterior distribution over possible Dynamic Bayesian Network structures. Our primary contributions are (i) a novel proposal distribution that efficiently samples sparse graphs and (ii) the relaxation of common restrictive modeling assumptions. We implement our method, named Sparse Signaling Pathway Sampling, in Julia using the Gen probabilistic programming language. Probabilistic programming is a powerful methodology for building statistical models. The resulting code is modular, extensible, and legible. The Gen language, in particular, allows us to customize our inference procedure for biological graphs and ensure efficient sampling. We evaluate our algorithm on simulated data and the HPN-DREAM pathway reconstruction challenge, comparing our performance against a variety of baseline methods. Our results demonstrate the vast potential for probabilistic programming, and Gen specifically, for biological network inference. Find the full codebase at https://github.com/gitter-lab/ssps
Unsupervised learning of transcriptional regulatory networks via latent tree graphical models
Sep 20, 2016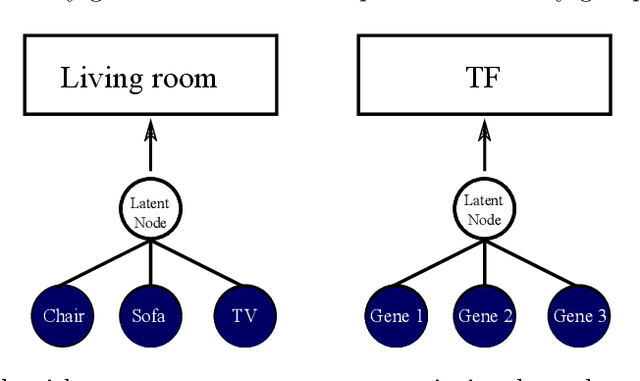
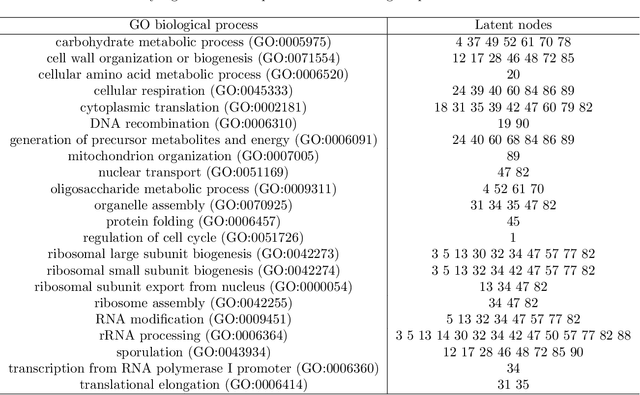
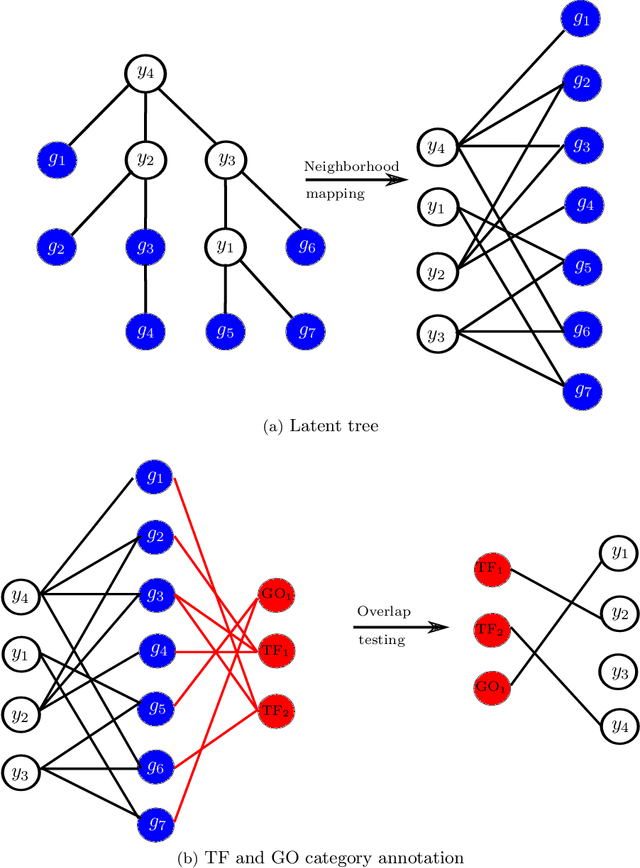
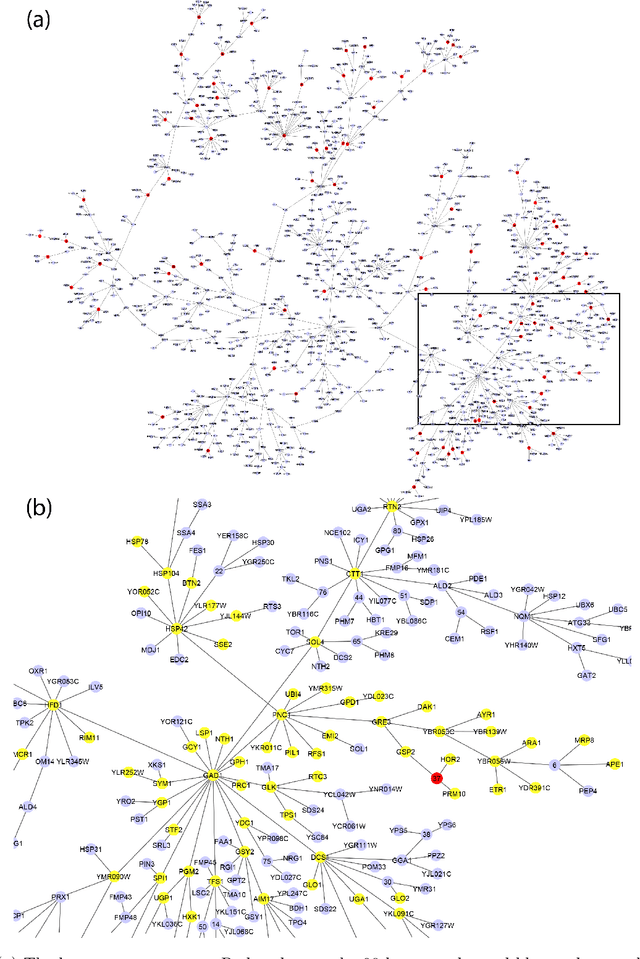
Gene expression is a readily-observed quantification of transcriptional activity and cellular state that enables the recovery of the relationships between regulators and their target genes. Reconstructing transcriptional regulatory networks from gene expression data is a problem that has attracted much attention, but previous work often makes the simplifying (but unrealistic) assumption that regulator activity is represented by mRNA levels. We use a latent tree graphical model to analyze gene expression without relying on transcription factor expression as a proxy for regulator activity. The latent tree model is a type of Markov random field that includes both observed gene variables and latent (hidden) variables, which factorize on a Markov tree. Through efficient unsupervised learning approaches, we determine which groups of genes are co-regulated by hidden regulators and the activity levels of those regulators. Post-processing annotates many of these discovered latent variables as specific transcription factors or groups of transcription factors. Other latent variables do not necessarily represent physical regulators but instead reveal hidden structure in the gene expression such as shared biological function. We apply the latent tree graphical model to a yeast stress response dataset. In addition to novel predictions, such as condition-specific binding of the transcription factor Msn4, our model recovers many known aspects of the yeast regulatory network. These include groups of co-regulated genes, condition-specific regulator activity, and combinatorial regulation among transcription factors. The latent tree graphical model is a general approach for analyzing gene expression data that requires no prior knowledge of which possible regulators exist, regulator activity, or where transcription factors physically bind.