 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrequency-Domain Latent Attention Gating for Cross-Domain Token Aggregation
Jun 06, 2026Token aggregation is a common bottleneck in models that map token representations to sample-level predictions, yet most pooling methods operate only in the original token domain. We propose FLaG, a plug-in aggregation module that transforms token representations with the real FFT, summarizes spectral components with learnable latent queries, applies a channel-wise gate, and reconstructs enhanced time-domain tokens for final pooling. We evaluate FLaG on antimicrobial peptide (AMP) activity prediction with ESM2, image classification with ResNet18 on CIFAR-10 and CIFAR-100, and text classification with RoBERTa on IMDB and GLUE. FLaG achieves its clearest gains on the ESM2-8M antimicrobial peptide tasks and on CIFAR-100, while remaining competitive with strong text baselines on IMDB and GLUE. Then we probe its behavior on the AMP setting with band knockouts, gate summaries, residue perturbations, latent-query readouts, and structure-proxy stratification. We find that low-frequency bands contribute the most overall, and the remaining higher-band pattern is more sample-specific. The gate acts as a broadly shared spectral reweighting stage and the cross-attention patterns are sample-specific with mild query-wise differentiation, and higher-helix peptides exhibit stronger average spectral sensitivity in both bacteria. The supplementary materials, source code and data are released at https://www.healthinformaticslab.org/supp/ and https://github.com/Kewei2023/AMPCliff/tree/FLaG.
A transformer-BiGRU-based framework with data augmentation and confident learning for network intrusion detection
Sep 05, 2025



In today's fast-paced digital communication, the surge in network traffic data and frequency demands robust and precise network intrusion solutions. Conventional machine learning methods struggle to grapple with complex patterns within the vast network intrusion datasets, which suffer from data scarcity and class imbalance. As a result, we have integrated machine learning and deep learning techniques within the network intrusion detection system to bridge this gap. This study has developed TrailGate, a novel framework that combines machine learning and deep learning techniques. By integrating Transformer and Bidirectional Gated Recurrent Unit (BiGRU) architectures with advanced feature selection strategies and supplemented by data augmentation techniques, TrailGate can identifies common attack types and excels at detecting and mitigating emerging threats. This algorithmic fusion excels at detecting common and well-understood attack types and has the unique ability to swiftly identify and neutralize emerging threats that stem from existing paradigms.
DeepSelective: Feature Gating and Representation Matching for Interpretable Clinical Prediction
Apr 15, 2025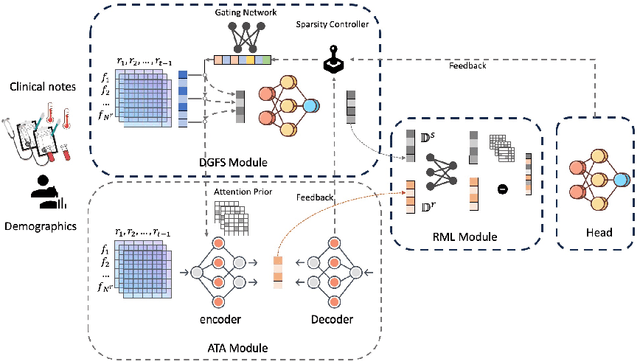

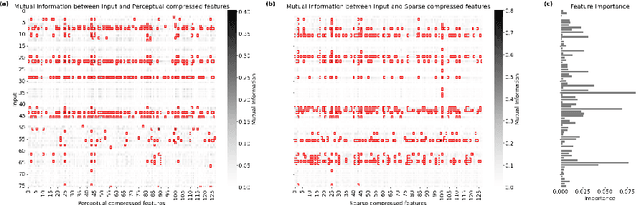
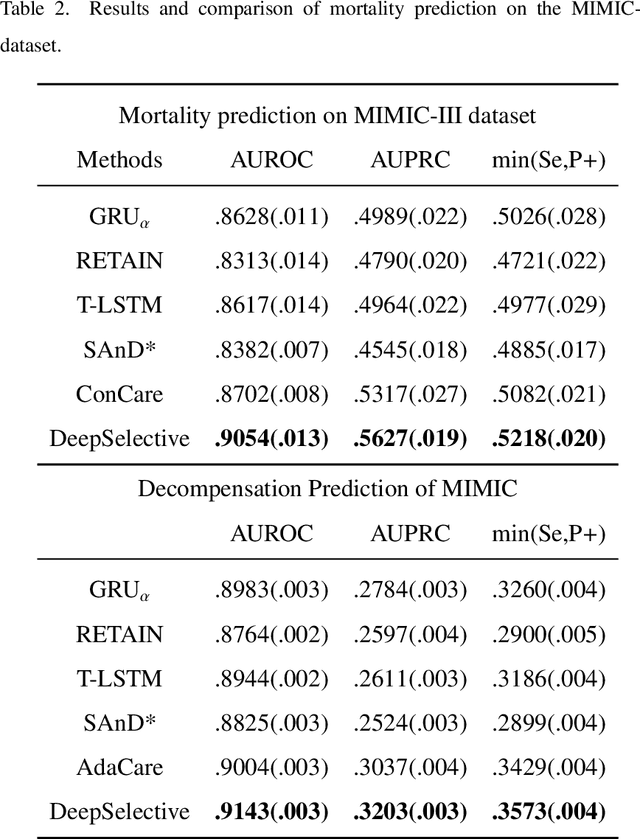
The rapid accumulation of Electronic Health Records (EHRs) has transformed healthcare by providing valuable data that enhance clinical predictions and diagnoses. While conventional machine learning models have proven effective, they often lack robust representation learning and depend heavily on expert-crafted features. Although deep learning offers powerful solutions, it is often criticized for its lack of interpretability. To address these challenges, we propose DeepSelective, a novel end to end deep learning framework for predicting patient prognosis using EHR data, with a strong emphasis on enhancing model interpretability. DeepSelective combines data compression techniques with an innovative feature selection approach, integrating custom-designed modules that work together to improve both accuracy and interpretability. Our experiments demonstrate that DeepSelective not only enhances predictive accuracy but also significantly improves interpretability, making it a valuable tool for clinical decision-making. The source code is freely available at http://www.healthinformaticslab.org/supp/resources.php .
Information Entropy Invariance: Enhancing Length Extrapolation in Attention Mechanisms
Jan 15, 2025Improving the length extrapolation capabilities of Large Language Models (LLMs) remains a critical challenge in natural language processing. Many recent efforts have focused on modifying the scaled dot-product attention mechanism, and often introduce scaled temperatures without rigorous theoretical justification. To fill this gap, we introduce a novel approach based on information entropy invariance. We propose two new scaled temperatures to enhance length extrapolation. First, a training-free method InfoScale is designed for dot-product attention, and preserves focus on original tokens during length extrapolation by ensuring information entropy remains consistent. Second, we theoretically analyze the impact of scaling (CosScale) on cosine attention. Experimental data demonstrates that combining InfoScale and CosScale achieves state-of-the-art performance on the GAU-{\alpha} model with a context window extended to 64 times the training length, and outperforms seven existing methods. Our analysis reveals that significantly increasing CosScale approximates windowed attention, and highlights the significance of attention score dilution as a key challenge in long-range context handling. The code and data are available at https://github.com/HT-NEKO/InfoScale.
AMPCliff: quantitative definition and benchmarking of activity cliffs in antimicrobial peptides
Apr 15, 2024



Activity cliff (AC) is a phenomenon that a pair of similar molecules differ by a small structural alternation but exhibit a large difference in their biochemical activities. The AC of small molecules has been extensively investigated but limited knowledge is accumulated about the AC phenomenon in peptides with canonical amino acids. This study introduces a quantitative definition and benchmarking framework AMPCliff for the AC phenomenon in antimicrobial peptides (AMPs) composed by canonical amino acids. A comprehensive analysis of the existing AMP dataset reveals a significant prevalence of AC within AMPs. AMPCliff quantifies the activities of AMPs by the metric minimum inhibitory concentration (MIC), and defines 0.9 as the minimum threshold for the normalized BLOSUM62 similarity score between a pair of aligned peptides with at least two-fold MIC changes. This study establishes a benchmark dataset of paired AMPs in Staphylococcus aureus from the publicly available AMP dataset GRAMPA, and conducts a rigorous procedure to evaluate various AMP AC prediction models, including nine machine learning, four deep learning algorithms, four masked language models, and four generative language models. Our analysis reveals that these models are capable of detecting AMP AC events and the pre-trained protein language ESM2 model demonstrates superior performance across the evaluations. The predictive performance of AMP activity cliffs remains to be further improved, considering that ESM2 with 33 layers only achieves the Spearman correlation coefficient=0.50 for the regression task of the MIC values on the benchmark dataset. Source code and additional resources are available at https://www.healthinformaticslab.org/supp/ or https://github.com/Kewei2023/AMPCliff-generation.
PepHarmony: A Multi-View Contrastive Learning Framework for Integrated Sequence and Structure-Based Peptide Encoding
Jan 21, 2024



Recent advances in protein language models have catalyzed significant progress in peptide sequence representation. Despite extensive exploration in this field, pre-trained models tailored for peptide-specific needs remain largely unaddressed due to the difficulty in capturing the complex and sometimes unstable structures of peptides. This study introduces a novel multi-view contrastive learning framework PepHarmony for the sequence-based peptide encoding task. PepHarmony innovatively combines both sequence- and structure-level information into a sequence-level encoding module through contrastive learning. We carefully select datasets from the Protein Data Bank (PDB) and AlphaFold database to encompass a broad spectrum of peptide sequences and structures. The experimental data highlights PepHarmony's exceptional capability in capturing the intricate relationship between peptide sequences and structures compared with the baseline and fine-tuned models. The robustness of our model is confirmed through extensive ablation studies, which emphasize the crucial roles of contrastive loss and strategic data sorting in enhancing predictive performance. The proposed PepHarmony framework serves as a notable contribution to peptide representations, and offers valuable insights for future applications in peptide drug discovery and peptide engineering. We have made all the source code utilized in this study publicly accessible via GitHub at https://github.com/zhangruochi/PepHarmony or http://www.healthinformaticslab.org/supp/.
PepLand: a large-scale pre-trained peptide representation model for a comprehensive landscape of both canonical and non-canonical amino acids
Nov 08, 2023In recent years, the scientific community has become increasingly interested on peptides with non-canonical amino acids due to their superior stability and resistance to proteolytic degradation. These peptides present promising modifications to biological, pharmacological, and physiochemical attributes in both endogenous and engineered peptides. Notwithstanding their considerable advantages, the scientific community exhibits a conspicuous absence of an effective pre-trained model adept at distilling feature representations from such complex peptide sequences. We herein propose PepLand, a novel pre-training architecture for representation and property analysis of peptides spanning both canonical and non-canonical amino acids. In essence, PepLand leverages a comprehensive multi-view heterogeneous graph neural network tailored to unveil the subtle structural representations of peptides. Empirical validations underscore PepLand's effectiveness across an array of peptide property predictions, encompassing protein-protein interactions, permeability, solubility, and synthesizability. The rigorous evaluation confirms PepLand's unparalleled capability in capturing salient synthetic peptide features, thereby laying a robust foundation for transformative advances in peptide-centric research domains. We have made all the source code utilized in this study publicly accessible via GitHub at https://github.com/zhangruochi/pepland
Hyper-SAGNN: a self-attention based graph neural network for hypergraphs
Nov 06, 2019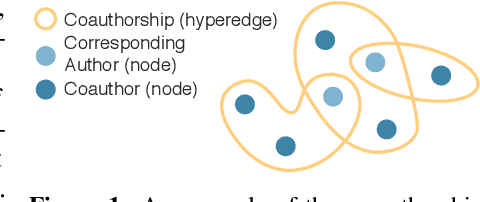

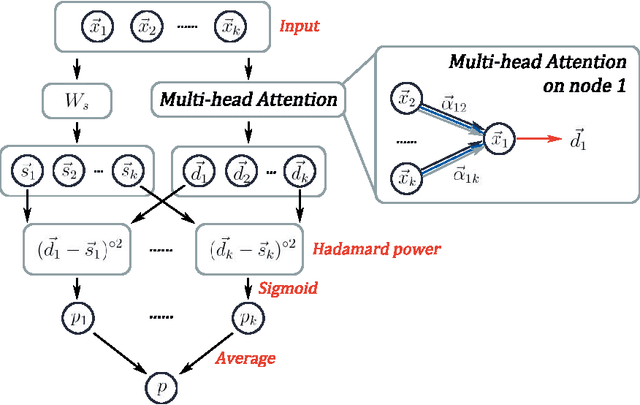
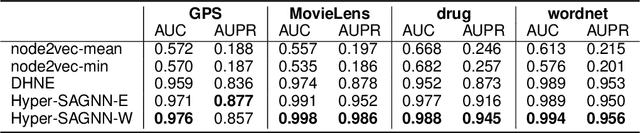
Graph representation learning for hypergraphs can be used to extract patterns among higher-order interactions that are critically important in many real world problems. Current approaches designed for hypergraphs, however, are unable to handle different types of hypergraphs and are typically not generic for various learning tasks. Indeed, models that can predict variable-sized heterogeneous hyperedges have not been available. Here we develop a new self-attention based graph neural network called Hyper-SAGNN applicable to homogeneous and heterogeneous hypergraphs with variable hyperedge sizes. We perform extensive evaluations on multiple datasets, including four benchmark network datasets and two single-cell Hi-C datasets in genomics. We demonstrate that Hyper-SAGNN significantly outperforms the state-of-the-art methods on traditional tasks while also achieving great performance on a new task called outsider identification. Hyper-SAGNN will be useful for graph representation learning to uncover complex higher-order interactions in different applications.