 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCan Current Agents Close the Discovery-to-Application Gap? A Case Study in Minecraft
Apr 27, 2026Discovering causal regularities and applying them to build functional systems--the discovery-to-application loop--is a hallmark of general intelligence, yet evaluating this capacity has been hindered by the vast complexity gap between scientific discovery and real-world engineering. We introduce SciCrafter, a Minecraft-based benchmark that operationalizes this loop through parameterized redstone circuit tasks. Agents must ignite lamps in specified patterns (e.g., simultaneously or in timed sequences); scaling target parameters substantially increases construction complexity and required knowledge, forcing genuine discovery rather than reliance on memorized solutions. Evaluating frontier models including GPT-5.2, Gemini-3-Pro, and Claude-Opus-4.5 under a general-purpose code agent scaffold, we find that all plateau at approximately 26% success rate. To diagnose these failures, we decompose the loop into four capacities--knowledge gap identification, experimental discovery, knowledge consolidation, and knowledge application--and design targeted interventions whose marginal contributions serve as proxies for corresponding gaps. Our analysis reveals that although the general knowledge application capability still remains as the biggest gap across all models, for frontier models the knowledge gap identification starts to become a major hurdle--indicating the bottleneck is shifting from solving problems right to raising the right problems for current AI. We release SciCrafter as a diagnostic probe for future research on AI systems that navigate the full discovery-to-application loop.
DMamba: Decomposition-enhanced Mamba for Time Series Forecasting
Feb 09, 2026State Space Models (SSMs), particularly Mamba, have shown potential in long-term time series forecasting. However, existing Mamba-based architectures often struggle with datasets characterized by non-stationary patterns. A key observation from time series theory is that the statistical nature of inter-variable relationships differs fundamentally between the trend and seasonal components of a decomposed series. Trend relationships are often driven by a few common stochastic factors or long-run equilibria, suggesting that they reside on a lower-dimensional manifold. In contrast, seasonal relationships involve dynamic, high-dimensional interactions like phase shifts and amplitude co-movements, requiring more expressive modeling. In this paper, we propose DMamba, a novel forecasting model that explicitly aligns architectural complexity with this component-specific characteristic. DMamba employs seasonal-trend decomposition and processes the components with specialized, differentially complex modules: a variable-direction Mamba encoder captures the rich, cross-variable dynamics within the seasonal component, while a simple Multi-Layer Perceptron (MLP) suffices to learn from the lower-dimensional inter-variable relationships in the trend component. Extensive experiments on diverse datasets demonstrate that DMamba sets a new state-of-the-art (SOTA), consistently outperforming both recent Mamba-based architectures and leading decomposition-based models.
FD-Bench: A Modular and Fair Benchmark for Data-driven Fluid Simulation
May 25, 2025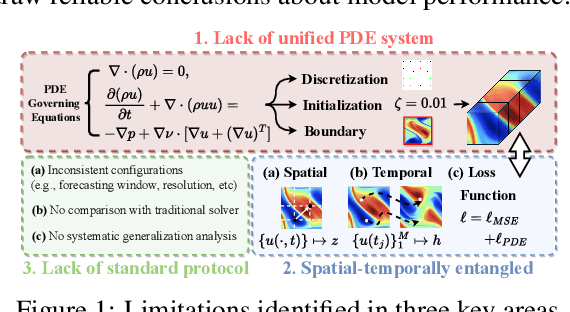

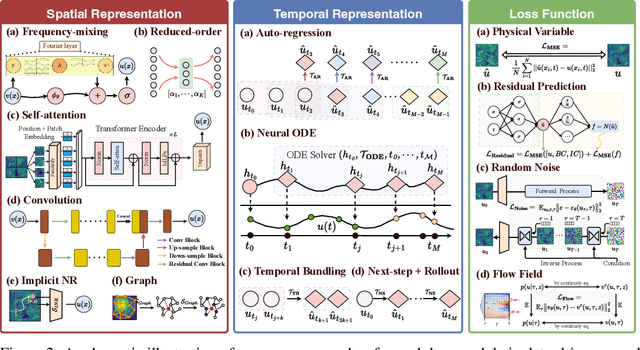
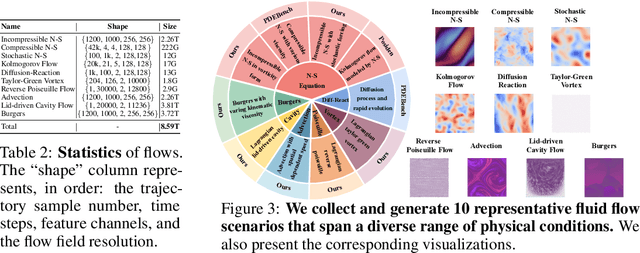
Data-driven modeling of fluid dynamics has advanced rapidly with neural PDE solvers, yet a fair and strong benchmark remains fragmented due to the absence of unified PDE datasets and standardized evaluation protocols. Although architectural innovations are abundant, fair assessment is further impeded by the lack of clear disentanglement between spatial, temporal and loss modules. In this paper, we introduce FD-Bench, the first fair, modular, comprehensive and reproducible benchmark for data-driven fluid simulation. FD-Bench systematically evaluates 85 baseline models across 10 representative flow scenarios under a unified experimental setup. It provides four key contributions: (1) a modular design enabling fair comparisons across spatial, temporal, and loss function modules; (2) the first systematic framework for direct comparison with traditional numerical solvers; (3) fine-grained generalization analysis across resolutions, initial conditions, and temporal windows; and (4) a user-friendly, extensible codebase to support future research. Through rigorous empirical studies, FD-Bench establishes the most comprehensive leaderboard to date, resolving long-standing issues in reproducibility and comparability, and laying a foundation for robust evaluation of future data-driven fluid models. The code is open-sourced at https://anonymous.4open.science/r/FD-Bench-15BC.
Graph Fourier Neural ODEs: Bridging Spatial and Temporal Multiscales in Molecular Dynamics
Nov 03, 2024



Molecular dynamics simulations are crucial for understanding complex physical, chemical, and biological processes at the atomic level. However, accurately capturing interactions across multiple spatial and temporal scales remains a significant challenge. We present a novel framework that jointly models spatial and temporal multiscale interactions in molecular dynamics. Our approach leverages Graph Fourier Transforms to decompose molecular structures into different spatial scales and employs Neural Ordinary Differential Equations to model the temporal dynamics in a curated manner influenced by the spatial modes. This unified framework links spatial structures with temporal evolution in a flexible manner, enabling more accurate and comprehensive simulations of molecular systems. We evaluate our model on the MD17 dataset, demonstrating consistent performance improvements over state-of-the-art baselines across multiple molecules, particularly under challenging conditions such as irregular timestep sampling and long-term prediction horizons. Ablation studies confirm the significant contributions of both spatial and temporal multiscale modeling components. Our method advances the simulation of complex molecular systems, potentially accelerating research in computational chemistry, drug discovery, and materials science.
Automated Molecular Concept Generation and Labeling with Large Language Models
Jun 13, 2024



Artificial intelligence (AI) is significantly transforming scientific research. Explainable AI methods, such as concept-based models (CMs), are promising for driving new scientific discoveries because they make predictions based on meaningful concepts and offer insights into the prediction process. In molecular science, however, explainable CMs are not as common compared to black-box models like Graph Neural Networks (GNNs), primarily due to their requirement for predefined concepts and manual label for each instance, which demand domain knowledge and can be labor-intensive. This paper introduces a novel framework for Automated Molecular Concept (AutoMolCo) generation and labeling. AutoMolCo leverages the knowledge in Large Language Models (LLMs) to automatically generate predictive molecular concepts and label them for each molecule. Such procedures are repeated through iterative interactions with LLMs to refine concepts, enabling simple linear models on the refined concepts to outperform GNNs and LLM in-context learning on several benchmarks. The whole AutoMolCo framework is automated without any human knowledge inputs in either concept generation, labeling, or refinement, thereby surpassing the limitations of extant CMs while maintaining their explainability and allowing easy intervention. Through systematic experiments on MoleculeNet and High-Throughput Experimentation (HTE) datasets, we demonstrate that the AutoMolCo-induced explainable CMs are beneficial and promising for molecular science research.
A Comprehensive Survey on Deep Graph Representation Learning
Apr 19, 2023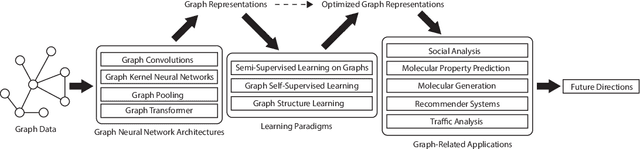
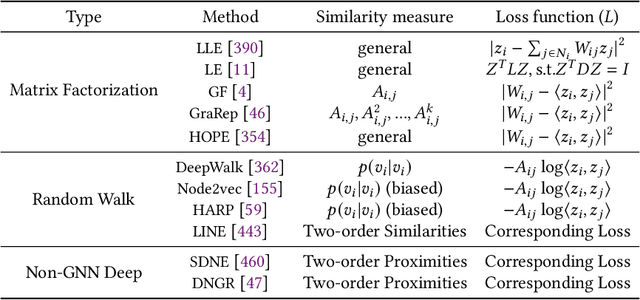


Graph representation learning aims to effectively encode high-dimensional sparse graph-structured data into low-dimensional dense vectors, which is a fundamental task that has been widely studied in a range of fields, including machine learning and data mining. Classic graph embedding methods follow the basic idea that the embedding vectors of interconnected nodes in the graph can still maintain a relatively close distance, thereby preserving the structural information between the nodes in the graph. However, this is sub-optimal due to: (i) traditional methods have limited model capacity which limits the learning performance; (ii) existing techniques typically rely on unsupervised learning strategies and fail to couple with the latest learning paradigms; (iii) representation learning and downstream tasks are dependent on each other which should be jointly enhanced. With the remarkable success of deep learning, deep graph representation learning has shown great potential and advantages over shallow (traditional) methods, there exist a large number of deep graph representation learning techniques have been proposed in the past decade, especially graph neural networks. In this survey, we conduct a comprehensive survey on current deep graph representation learning algorithms by proposing a new taxonomy of existing state-of-the-art literature. Specifically, we systematically summarize the essential components of graph representation learning and categorize existing approaches by the ways of graph neural network architectures and the most recent advanced learning paradigms. Moreover, this survey also provides the practical and promising applications of deep graph representation learning. Last but not least, we state new perspectives and suggest challenging directions which deserve further investigations in the future.
DisenPOI: Disentangling Sequential and Geographical Influence for Point-of-Interest Recommendation
Oct 29, 2022



Point-of-Interest (POI) recommendation plays a vital role in various location-aware services. It has been observed that POI recommendation is driven by both sequential and geographical influences. However, since there is no annotated label of the dominant influence during recommendation, existing methods tend to entangle these two influences, which may lead to sub-optimal recommendation performance and poor interpretability. In this paper, we address the above challenge by proposing DisenPOI, a novel Disentangled dual-graph framework for POI recommendation, which jointly utilizes sequential and geographical relationships on two separate graphs and disentangles the two influences with self-supervision. The key novelty of our model compared with existing approaches is to extract disentangled representations of both sequential and geographical influences with contrastive learning. To be specific, we construct a geographical graph and a sequential graph based on the check-in sequence of a user. We tailor their propagation schemes to become sequence-/geo-aware to better capture the corresponding influences. Preference proxies are extracted from check-in sequence as pseudo labels for the two influences, which supervise the disentanglement via a contrastive loss. Extensive experiments on three datasets demonstrate the superiority of the proposed model.
Over-smoothing Effect of Graph Convolutional Networks
Feb 01, 2022

Over-smoothing is a severe problem which limits the depth of Graph Convolutional Networks. This article gives a comprehensive analysis of the mechanism behind Graph Convolutional Networks and the over-smoothing effect. The article proposes an upper bound for the occurrence of over-smoothing, which offers insight into the key factors behind over-smoothing. The results presented in this article successfully explain the feasibility of several algorithms that alleviate over-smoothing.
Minimal Solutions for Relative Pose with a Single Affine Correspondence
Dec 23, 2019
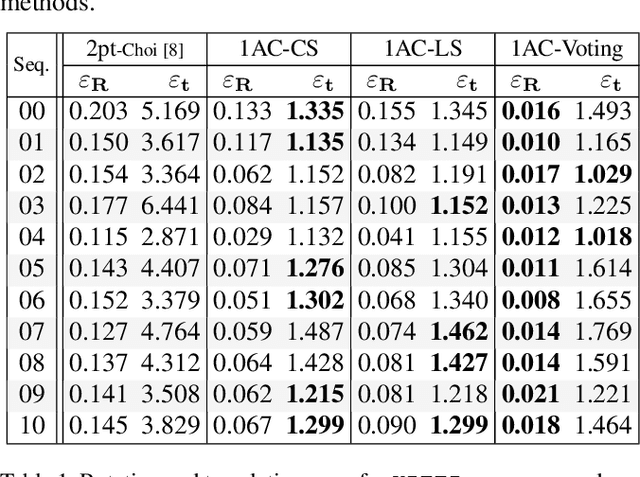
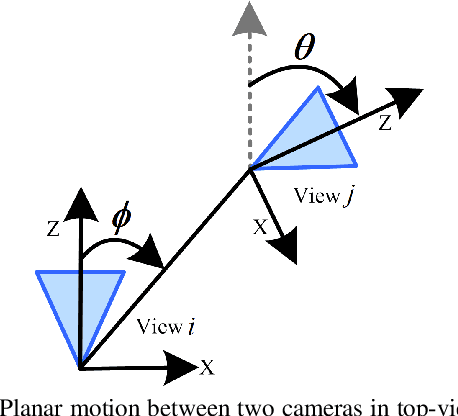
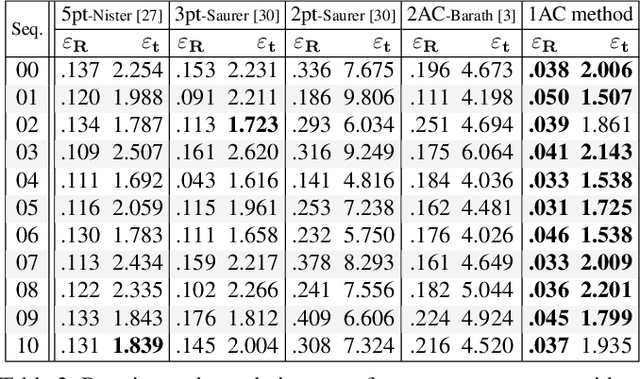
In this paper we present four cases of minimal solutions for two-view relative pose estimation by exploiting the affine transformation between feature points and we demonstrate efficient solvers for these cases. It is shown, that under the planar motion assumption or with knowledge of a vertical direction, a single affine correspondence is sufficient to recover the relative camera pose. The four cases considered are two-view planar relative motion for calibrated cameras as a closed-form and a least-squares solution, a closed-form solution for unknown focal length and the case of a known vertical direction. These algorithms can be used efficiently for outlier detection within a RANSAC loop and for initial motion estimation. All the methods are evaluated on both synthetic data and real-world datasets from the KITTI benchmark. The experimental results demonstrate that our methods outperform comparable state-of-the-art methods in accuracy with the benefit of a reduced number of needed RANSAC iterations.