 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTeam of Thoughts: Efficient Test-time Scaling of Agentic Systems through Orchestrated Tool Calling
Feb 18, 2026Existing Multi-Agent Systems (MAS) typically rely on static, homogeneous model configurations, limiting their ability to exploit the distinct strengths of differently post-trained models. To address this, we introduce Team-of-Thoughts, a novel MAS architecture that leverages the complementary capabilities of heterogeneous agents via an orchestrator-tool paradigm. Our framework introduces two key mechanisms to optimize performance: (1) an orchestrator calibration scheme that identifies models with superior coordination capabilities, and (2) a self-assessment protocol where tool agents profile their own domain expertise to account for variations in post-training skills. During inference, the orchestrator dynamically activates the most suitable tool agents based on these proficiency profiles. Experiments on five reasoning and code generation benchmarks show that Team-of-Thoughts delivers consistently superior task performance. Notably, on AIME24 and LiveCodeBench, our approach achieves accuracies of 96.67% and 72.53%, respectively, substantially outperforming homogeneous role-play baselines, which score 80% and 65.93%.
Deep Kernel Fusion for Transformers
Feb 12, 2026Agentic LLM inference with long contexts is increasingly limited by memory bandwidth rather than compute. In this setting, SwiGLU MLP blocks, whose large weights exceed cache capacity, become a major yet under-optimized bottleneck. We propose DeepFusionKernel, a deeply fused kernel that cuts HBM traffic and boosts cache reuse, delivering up to 13.2% speedup on H100 and 9.7% on A100 over SGLang. Integrated with SGLang and paired with a kernel scheduler, DeepFusionKernel ensures consistent accelerations over generation lengths, while remaining adaptable to diverse models, inference configurations, and hardware platforms.
Heterogeneous Computing: The Key to Powering the Future of AI Agent Inference
Jan 29, 2026AI agent inference is driving an inference heavy datacenter future and exposes bottlenecks beyond compute - especially memory capacity, memory bandwidth and high-speed interconnect. We introduce two metrics - Operational Intensity (OI) and Capacity Footprint (CF) - that jointly explain regimes the classic roofline analysis misses, including the memory capacity wall. Across agentic workflows (chat, coding, web use, computer use) and base model choices (GQA/MLA, MoE, quantization), OI/CF can shift dramatically, with long context KV cache making decode highly memory bound. These observations motivate disaggregated serving and system level heterogeneity: specialized prefill and decode accelerators, broader scale up networking, and decoupled compute-memory enabled by optical I/O. We further hypothesize agent-hardware co design, multiple inference accelerators within one system, and high bandwidth, large capacity memory disaggregation as foundations for adaptation to evolving OI/CF. Together, these directions chart a path to sustain efficiency and capability for large scale agentic AI inference.
CaMeLs Can Use Computers Too: System-level Security for Computer Use Agents
Jan 14, 2026AI agents are vulnerable to prompt injection attacks, where malicious content hijacks agent behavior to steal credentials or cause financial loss. The only known robust defense is architectural isolation that strictly separates trusted task planning from untrusted environment observations. However, applying this design to Computer Use Agents (CUAs) -- systems that automate tasks by viewing screens and executing actions -- presents a fundamental challenge: current agents require continuous observation of UI state to determine each action, conflicting with the isolation required for security. We resolve this tension by demonstrating that UI workflows, while dynamic, are structurally predictable. We introduce Single-Shot Planning for CUAs, where a trusted planner generates a complete execution graph with conditional branches before any observation of potentially malicious content, providing provable control flow integrity guarantees against arbitrary instruction injections. Although this architectural isolation successfully prevents instruction injections, we show that additional measures are needed to prevent Branch Steering attacks, which manipulate UI elements to trigger unintended valid paths within the plan. We evaluate our design on OSWorld, and retain up to 57% of the performance of frontier models while improving performance for smaller open-source models by up to 19%, demonstrating that rigorous security and utility can coexist in CUAs.
On the Existence and Behaviour of Secondary Attention Sinks
Dec 22, 2025Attention sinks are tokens, often the beginning-of-sequence (BOS) token, that receive disproportionately high attention despite limited semantic relevance. In this work, we identify a class of attention sinks, which we term secondary sinks, that differ fundamentally from the sinks studied in prior works, which we term primary sinks. While prior works have identified that tokens other than BOS can sometimes become sinks, they were found to exhibit properties analogous to the BOS token. Specifically, they emerge at the same layer, persist throughout the network and draw a large amount of attention mass. Whereas, we find the existence of secondary sinks that arise primarily in middle layers and can persist for a variable number of layers, and draw a smaller, but still significant, amount of attention mass. Through extensive experiments across 11 model families, we analyze where these secondary sinks appear, their properties, how they are formed, and their impact on the attention mechanism. Specifically, we show that: (1) these sinks are formed by specific middle-layer MLP modules; these MLPs map token representations to vectors that align with the direction of the primary sink of that layer. (2) The $\ell_2$-norm of these vectors determines the sink score of the secondary sink, and also the number of layers it lasts for, thereby leading to different impacts on the attention mechanisms accordingly. (3) The primary sink weakens in middle layers, coinciding with the emergence of secondary sinks. We observe that in larger-scale models, the location and lifetime of the sinks, together referred to as sink levels, appear in a more deterministic and frequent manner. Specifically, we identify three sink levels in QwQ-32B and six levels in Qwen3-14B.
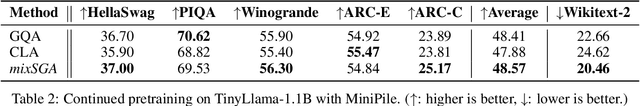
Mixture of Weight-shared Heterogeneous Group Attention Experts for Dynamic Token-wise KV Optimization
Jun 16, 2025
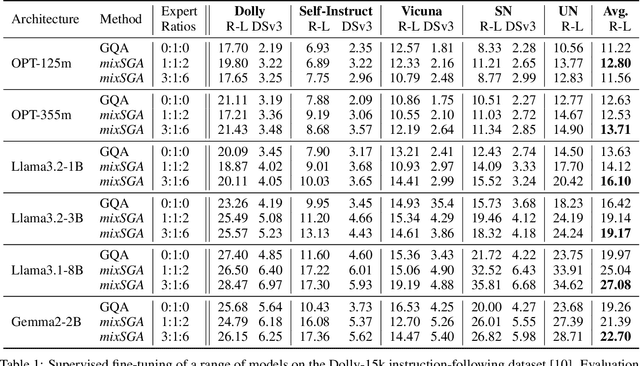
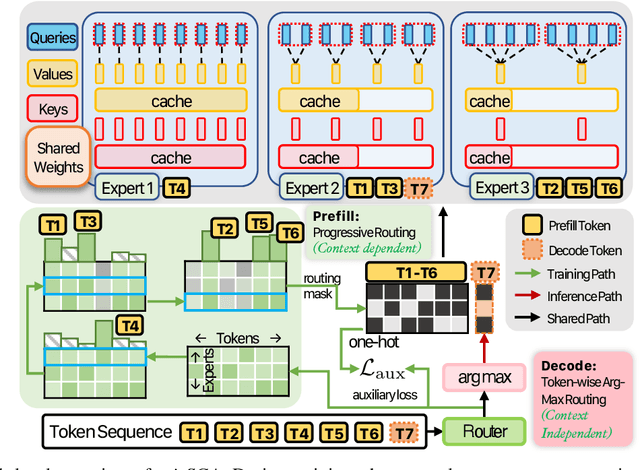
Transformer models face scalability challenges in causal language modeling (CLM) due to inefficient memory allocation for growing key-value (KV) caches, which strains compute and storage resources. Existing methods like Grouped Query Attention (GQA) and token-level KV optimization improve efficiency but rely on rigid resource allocation, often discarding "low-priority" tokens or statically grouping them, failing to address the dynamic spectrum of token importance. We propose mixSGA, a novel mixture-of-expert (MoE) approach that dynamically optimizes token-wise computation and memory allocation. Unlike prior approaches, mixSGA retains all tokens while adaptively routing them to specialized experts with varying KV group sizes, balancing granularity and efficiency. Our key novelties include: (1) a token-wise expert-choice routing mechanism guided by learned importance scores, enabling proportional resource allocation without token discard; (2) weight-sharing across grouped attention projections to minimize parameter overhead; and (3) an auxiliary loss to ensure one-hot routing decisions for training-inference consistency in CLMs. Extensive evaluations across Llama3, TinyLlama, OPT, and Gemma2 model families show mixSGA's superiority over static baselines. On instruction-following and continued pretraining tasks, mixSGA achieves higher ROUGE-L and lower perplexity under the same KV budgets.
A3 : an Analytical Low-Rank Approximation Framework for Attention
May 19, 2025Large language models have demonstrated remarkable performance; however, their massive parameter counts make deployment highly expensive. Low-rank approximation offers a promising compression solution, yet existing approaches have two main limitations: (1) They focus on minimizing the output error of individual linear layers, without considering the architectural characteristics of Transformers, and (2) they decompose a large weight matrix into two small low-rank matrices. Consequently, these methods often fall short compared to other compression techniques like pruning and quantization, and introduce runtime overhead such as the extra GEMM kernel launches for decomposed small matrices. To address these limitations, we propose $\tt A^\tt 3$, a post-training low-rank approximation framework. $\tt A^\tt 3$ splits a Transformer layer into three functional components, namely $\tt QK$, $\tt OV$, and $\tt MLP$. For each component, $\tt A^\tt 3$ provides an analytical solution that reduces the hidden dimension size inside each component while minimizing the component's functional loss ($\it i.e.$, error in attention scores, attention outputs, and MLP outputs). This approach directly reduces model sizes, KV cache sizes, and FLOPs without introducing any runtime overheads. In addition, it provides a new narrative in advancing the optimization problem from singular linear layer loss optimization toward improved end-to-end performance. Through extensive experiments, we show that $\tt A^\tt 3$ maintains superior performance compared to SoTAs. For example, under the same reduction budget in computation and memory, our low-rank approximated LLaMA 3.1-70B achieves a perplexity of 4.69 on WikiText-2, outperforming the previous SoTA's 7.87 by 3.18. We also demonstrate the versatility of $\tt A^\tt 3$, including KV cache compression, quantization, and mixed-rank assignments for enhanced performance.
ARIES: Autonomous Reasoning with LLMs on Interactive Thought Graph Environments
Feb 28, 2025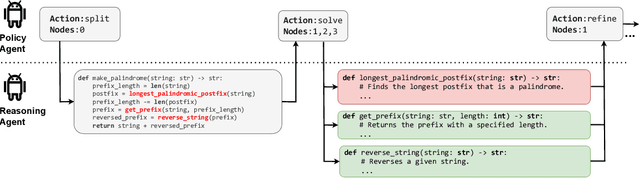



Recent research has shown that LLM performance on reasoning tasks can be enhanced by scaling test-time compute. One promising approach, particularly with decomposable problems, involves arranging intermediate solutions as a graph on which transformations are performed to explore the solution space. However, prior works rely on pre-determined, task-specific transformation schedules which are subject to a set of searched hyperparameters. In this work, we view thought graph transformations as actions in a Markov decision process, and implement policy agents to drive effective action policies for the underlying reasoning LLM agent. In particular, we investigate the ability for another LLM to act as a policy agent on thought graph environments and introduce ARIES, a multi-agent architecture for reasoning with LLMs. In ARIES, reasoning LLM agents solve decomposed subproblems, while policy LLM agents maintain visibility of the thought graph states, and dynamically adapt the problem-solving strategy. Through extensive experiments, we observe that using off-the-shelf LLMs as policy agents with no supervised fine-tuning (SFT) can yield up to $29\%$ higher accuracy on HumanEval relative to static transformation schedules, as well as reducing inference costs by $35\%$ and avoid any search requirements. We also conduct a thorough analysis of observed failure modes, highlighting that limitations on LLM sizes and the depth of problem decomposition can be seen as challenges to scaling LLM-guided reasoning.
AMPLE: Event-Driven Accelerator for Mixed-Precision Inference of Graph Neural Networks
Feb 28, 2025
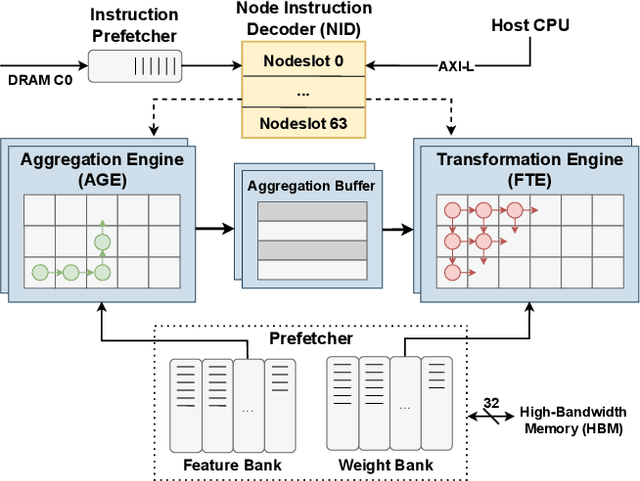

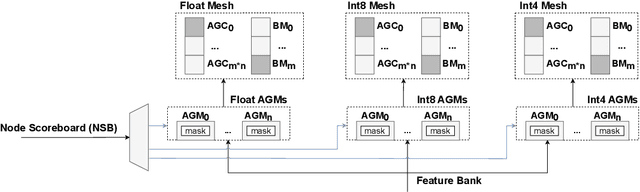
Graph Neural Networks (GNNs) have recently gained attention due to their performance on non-Euclidean data. The use of custom hardware architectures proves particularly beneficial for GNNs due to their irregular memory access patterns, resulting from the sparse structure of graphs. However, existing FPGA accelerators are limited by their double buffering mechanism, which doesn't account for the irregular node distribution in typical graph datasets. To address this, we introduce \textbf{AMPLE} (Accelerated Message Passing Logic Engine), an FPGA accelerator leveraging a new event-driven programming flow. We develop a mixed-arithmetic architecture, enabling GNN inference to be quantized at a node-level granularity. Finally, prefetcher for data and instructions is implemented to optimize off-chip memory access and maximize node parallelism. Evaluation on citation and social media graph datasets ranging from $2$K to $700$K nodes showed a mean speedup of $243\times$ and $7.2\times$ against CPU and GPU counterparts, respectively.
Cached Multi-Lora Composition for Multi-Concept Image Generation
Feb 07, 2025Low-Rank Adaptation (LoRA) has emerged as a widely adopted technique in text-to-image models, enabling precise rendering of multiple distinct elements, such as characters and styles, in multi-concept image generation. However, current approaches face significant challenges when composing these LoRAs for multi-concept image generation, resulting in diminished generated image quality. In this paper, we initially investigate the role of LoRAs in the denoising process through the lens of the Fourier frequency domain. Based on the hypothesis that applying multiple LoRAs could lead to "semantic conflicts", we find that certain LoRAs amplify high-frequency features such as edges and textures, whereas others mainly focus on low-frequency elements, including the overall structure and smooth color gradients. Building on these insights, we devise a frequency domain based sequencing strategy to determine the optimal order in which LoRAs should be integrated during inference. This strategy offers a methodical and generalizable solution compared to the naive integration commonly found in existing LoRA fusion techniques. To fully leverage our proposed LoRA order sequence determination method in multi-LoRA composition tasks, we introduce a novel, training-free framework, Cached Multi-LoRA (CMLoRA), designed to efficiently integrate multiple LoRAs while maintaining cohesive image generation. With its flexible backbone for multi-LoRA fusion and a non-uniform caching strategy tailored to individual LoRAs, CMLoRA has the potential to reduce semantic conflicts in LoRA composition and improve computational efficiency. Our experimental evaluations demonstrate that CMLoRA outperforms state-of-the-art training-free LoRA fusion methods by a significant margin -- it achieves an average improvement of $2.19\%$ in CLIPScore, and $11.25\%$ in MLLM win rate compared to LoraHub, LoRA Composite, and LoRA Switch.