 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOmni-DNA: A Unified Genomic Foundation Model for Cross-Modal and Multi-Task Learning
Feb 05, 2025



Large Language Models (LLMs) demonstrate remarkable generalizability across diverse tasks, yet genomic foundation models (GFMs) still require separate finetuning for each downstream application, creating significant overhead as model sizes grow. Moreover, existing GFMs are constrained by rigid output formats, limiting their applicability to various genomic tasks. In this work, we revisit the transformer-based auto-regressive models and introduce Omni-DNA, a family of cross-modal multi-task models ranging from 20 million to 1 billion parameters. Our approach consists of two stages: (i) pretraining on DNA sequences with next token prediction objective, and (ii) expanding the multi-modal task-specific tokens and finetuning for multiple downstream tasks simultaneously. When evaluated on the Nucleotide Transformer and GB benchmarks, Omni-DNA achieves state-of-the-art performance on 18 out of 26 tasks. Through multi-task finetuning, Omni-DNA addresses 10 acetylation and methylation tasks at once, surpassing models trained on each task individually. Finally, we design two complex genomic tasks, DNA2Function and Needle-in-DNA, which map DNA sequences to textual functional descriptions and images, respectively, indicating Omni-DNA's cross-modal capabilities to broaden the scope of genomic applications. All the models are available through https://huggingface.co/collections/zehui127
Absorb & Escape: Overcoming Single Model Limitations in Generating Genomic Sequences
Oct 28, 2024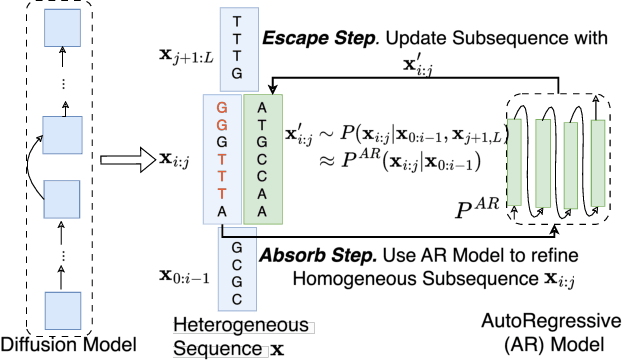


Abstract Recent advances in immunology and synthetic biology have accelerated the development of deep generative methods for DNA sequence design. Two dominant approaches in this field are AutoRegressive (AR) models and Diffusion Models (DMs). However, genomic sequences are functionally heterogeneous, consisting of multiple connected regions (e.g., Promoter Regions, Exons, and Introns) where elements within each region come from the same probability distribution, but the overall sequence is non-homogeneous. This heterogeneous nature presents challenges for a single model to accurately generate genomic sequences. In this paper, we analyze the properties of AR models and DMs in heterogeneous genomic sequence generation, pointing out crucial limitations in both methods: (i) AR models capture the underlying distribution of data by factorizing and learning the transition probability but fail to capture the global property of DNA sequences. (ii) DMs learn to recover the global distribution but tend to produce errors at the base pair level. To overcome the limitations of both approaches, we propose a post-training sampling method, termed Absorb & Escape (A&E) to perform compositional generation from AR models and DMs. This approach starts with samples generated by DMs and refines the sample quality using an AR model through the alternation of the Absorb and Escape steps. To assess the quality of generated sequences, we conduct extensive experiments on 15 species for conditional and unconditional DNA generation. The experiment results from motif distribution, diversity checks, and genome integration tests unequivocally show that A&E outperforms state-of-the-art AR models and DMs in genomic sequence generation.
GV-Rep: A Large-Scale Dataset for Genetic Variant Representation Learning
Jul 24, 2024


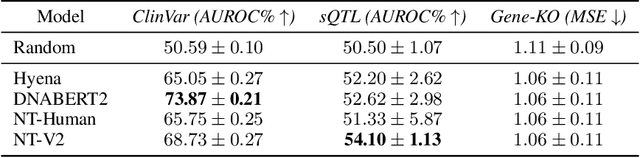
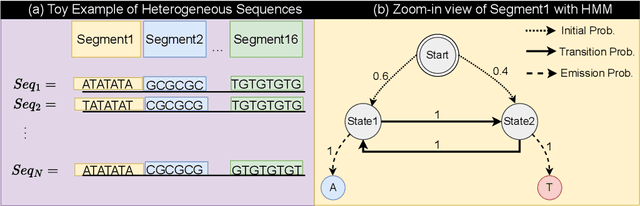
Genetic variants (GVs) are defined as differences in the DNA sequences among individuals and play a crucial role in diagnosing and treating genetic diseases. The rapid decrease in next generation sequencing cost has led to an exponential increase in patient-level GV data. This growth poses a challenge for clinicians who must efficiently prioritize patient-specific GVs and integrate them with existing genomic databases to inform patient management. To addressing the interpretation of GVs, genomic foundation models (GFMs) have emerged. However, these models lack standardized performance assessments, leading to considerable variability in model evaluations. This poses the question: How effectively do deep learning methods classify unknown GVs and align them with clinically-verified GVs? We argue that representation learning, which transforms raw data into meaningful feature spaces, is an effective approach for addressing both indexing and classification challenges. We introduce a large-scale Genetic Variant dataset, named GV-Rep, featuring variable-length contexts and detailed annotations, designed for deep learning models to learn GV representations across various traits, diseases, tissue types, and experimental contexts. Our contributions are three-fold: (i) Construction of a comprehensive dataset with 7 million records, each labeled with characteristics of the corresponding variants, alongside additional data from 17,548 gene knockout tests across 1,107 cell types, 1,808 variant combinations, and 156 unique clinically verified GVs from real-world patients. (ii) Analysis of the structure and properties of the dataset. (iii) Experimentation of the dataset with pre-trained GFMs. The results show a significant gap between GFMs current capabilities and accurate GV representation. We hope this dataset will help advance genomic deep learning to bridge this gap.
Enhancing Real-World Complex Network Representations with Hyperedge Augmentation
Feb 20, 2024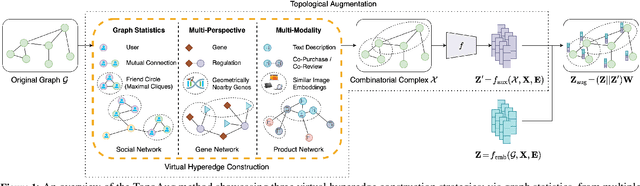
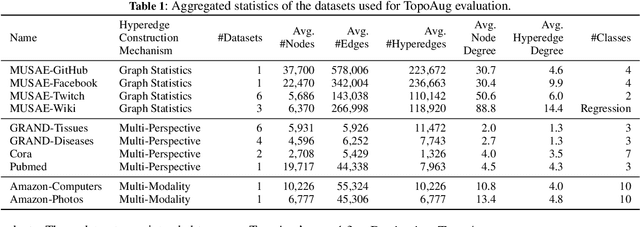
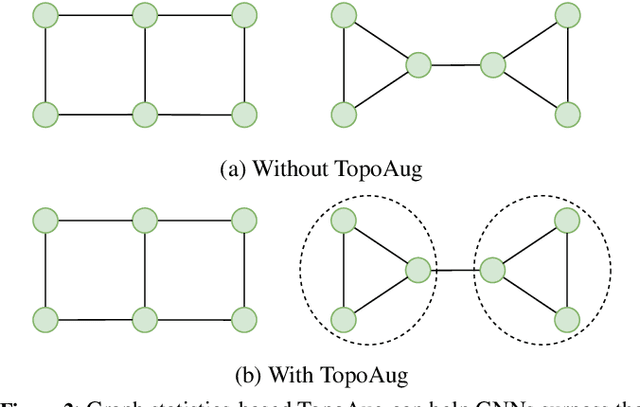
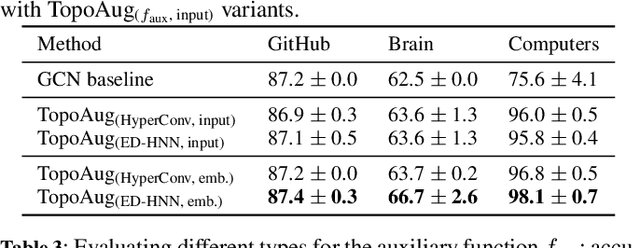
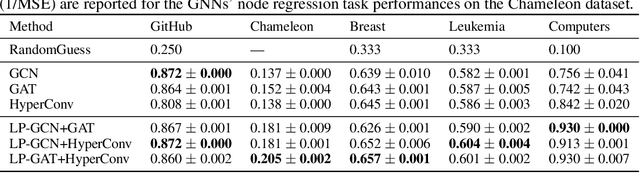
Graph augmentation methods play a crucial role in improving the performance and enhancing generalisation capabilities in Graph Neural Networks (GNNs). Existing graph augmentation methods mainly perturb the graph structures and are usually limited to pairwise node relations. These methods cannot fully address the complexities of real-world large-scale networks that often involve higher-order node relations beyond only being pairwise. Meanwhile, real-world graph datasets are predominantly modelled as simple graphs, due to the scarcity of data that can be used to form higher-order edges. Therefore, reconfiguring the higher-order edges as an integration into graph augmentation strategies lights up a promising research path to address the aforementioned issues. In this paper, we present Hyperedge Augmentation (HyperAug), a novel graph augmentation method that constructs virtual hyperedges directly form the raw data, and produces auxiliary node features by extracting from the virtual hyperedge information, which are used for enhancing GNN performances on downstream tasks. We design three diverse virtual hyperedge construction strategies to accompany the augmentation scheme: (1) via graph statistics, (2) from multiple data perspectives, and (3) utilising multi-modality. Furthermore, to facilitate HyperAug evaluation, we provide 23 novel real-world graph datasets across various domains including social media, biology, and e-commerce. Our empirical study shows that HyperAug consistently and significantly outperforms GNN baselines and other graph augmentation methods, across a variety of application contexts, which clearly indicates that it can effectively incorporate higher-order node relations into graph augmentation methods for real-world complex networks.
DiscDiff: Latent Diffusion Model for DNA Sequence Generation
Feb 08, 2024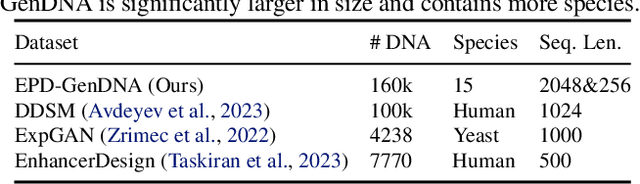
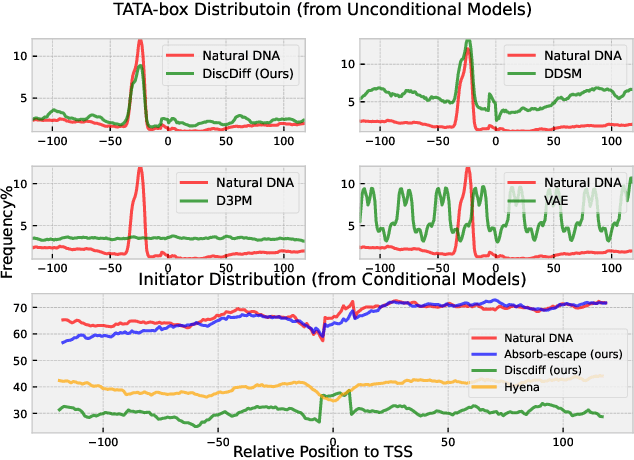

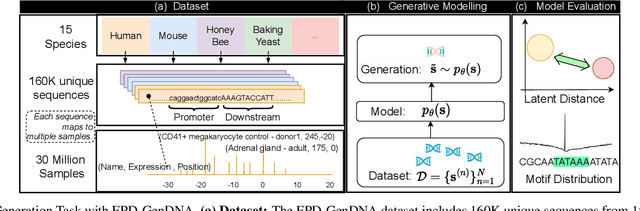
This paper introduces a novel framework for DNA sequence generation, comprising two key components: DiscDiff, a Latent Diffusion Model (LDM) tailored for generating discrete DNA sequences, and Absorb-Escape, a post-training algorithm designed to refine these sequences. Absorb-Escape enhances the realism of the generated sequences by correcting `round errors' inherent in the conversion process between latent and input spaces. Our approach not only sets new standards in DNA sequence generation but also demonstrates superior performance over existing diffusion models, in generating both short and long DNA sequences. Additionally, we introduce EPD-GenDNA, the first comprehensive, multi-species dataset for DNA generation, encompassing 160,000 unique sequences from 15 species. We hope this study will advance the generative modelling of DNA, with potential implications for gene therapy and protein production.
Latent Diffusion Model for DNA Sequence Generation
Oct 09, 2023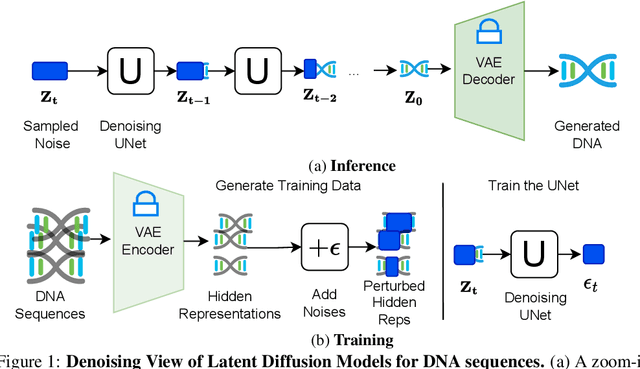
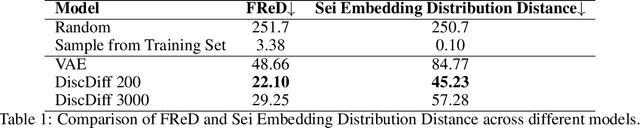
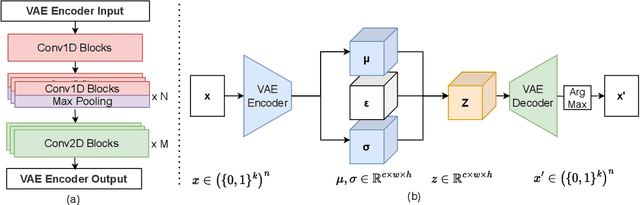
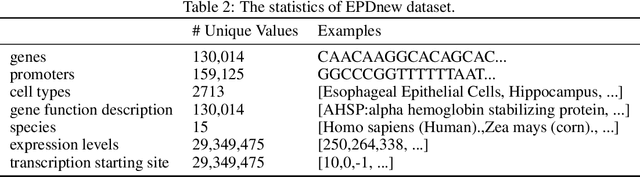
The harnessing of machine learning, especially deep generative models, has opened up promising avenues in the field of synthetic DNA sequence generation. Whilst Generative Adversarial Networks (GANs) have gained traction for this application, they often face issues such as limited sample diversity and mode collapse. On the other hand, Diffusion Models are a promising new class of generative models that are not burdened with these problems, enabling them to reach the state-of-the-art in domains such as image generation. In light of this, we propose a novel latent diffusion model, DiscDiff, tailored for discrete DNA sequence generation. By simply embedding discrete DNA sequences into a continuous latent space using an autoencoder, we are able to leverage the powerful generative abilities of continuous diffusion models for the generation of discrete data. Additionally, we introduce Fr\'echet Reconstruction Distance (FReD) as a new metric to measure the sample quality of DNA sequence generations. Our DiscDiff model demonstrates an ability to generate synthetic DNA sequences that align closely with real DNA in terms of Motif Distribution, Latent Embedding Distribution (FReD), and Chromatin Profiles. Additionally, we contribute a comprehensive cross-species dataset of 150K unique promoter-gene sequences from 15 species, enriching resources for future generative modelling in genomics. We will make our code public upon publication.
Genomic Interpreter: A Hierarchical Genomic Deep Neural Network with 1D Shifted Window Transformer
Jun 28, 2023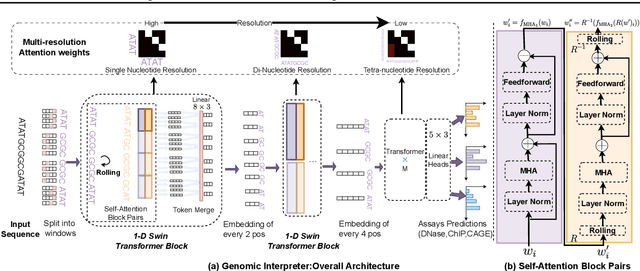
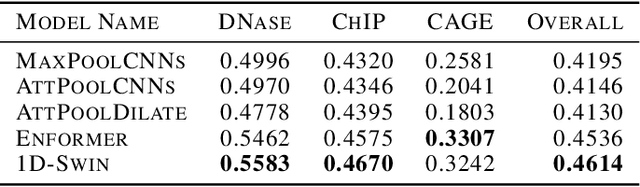
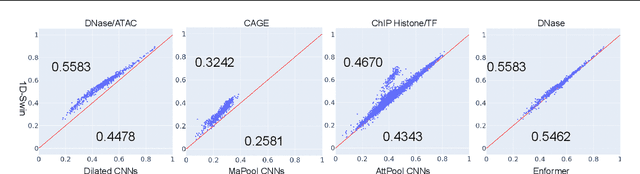
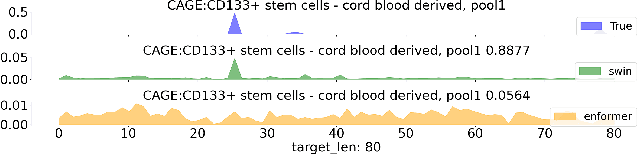
Given the increasing volume and quality of genomics data, extracting new insights requires interpretable machine-learning models. This work presents Genomic Interpreter: a novel architecture for genomic assay prediction. This model outperforms the state-of-the-art models for genomic assay prediction tasks. Our model can identify hierarchical dependencies in genomic sites. This is achieved through the integration of 1D-Swin, a novel Transformer-based block designed by us for modelling long-range hierarchical data. Evaluated on a dataset containing 38,171 DNA segments of 17K base pairs, Genomic Interpreter demonstrates superior performance in chromatin accessibility and gene expression prediction and unmasks the underlying `syntax' of gene regulation.
Hybrid Graph: A Unified Graph Representation with Datasets and Benchmarks for Complex Graphs
Jun 08, 2023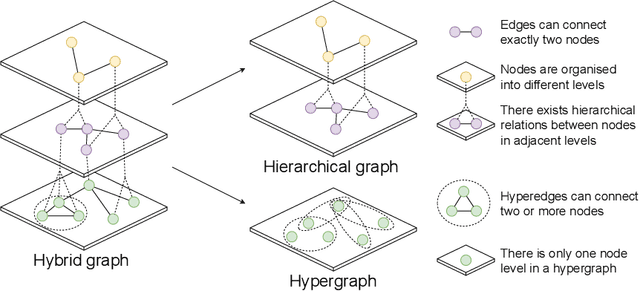
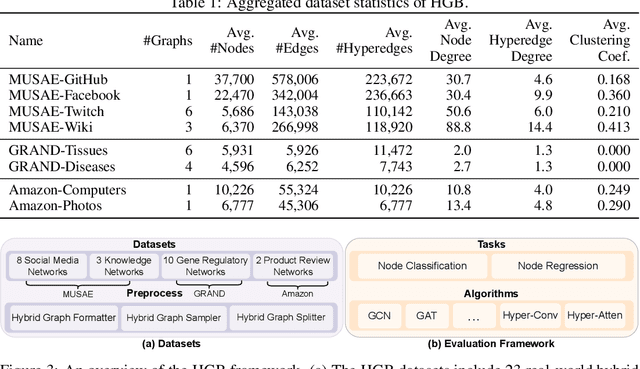
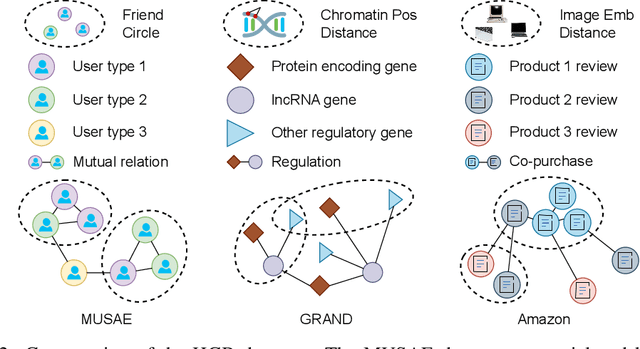
Graphs are widely used to encapsulate a variety of data formats, but real-world networks often involve complex node relations beyond only being pairwise. While hypergraphs and hierarchical graphs have been developed and employed to account for the complex node relations, they cannot fully represent these complexities in practice. Additionally, though many Graph Neural Networks (GNNs) have been proposed for representation learning on higher-order graphs, they are usually only evaluated on simple graph datasets. Therefore, there is a need for a unified modelling of higher-order graphs, and a collection of comprehensive datasets with an accessible evaluation framework to fully understand the performance of these algorithms on complex graphs. In this paper, we introduce the concept of hybrid graphs, a unified definition for higher-order graphs, and present the Hybrid Graph Benchmark (HGB). HGB contains 23 real-world hybrid graph datasets across various domains such as biology, social media, and e-commerce. Furthermore, we provide an extensible evaluation framework and a supporting codebase to facilitate the training and evaluation of GNNs on HGB. Our empirical study of existing GNNs on HGB reveals various research opportunities and gaps, including (1) evaluating the actual performance improvement of hypergraph GNNs over simple graph GNNs; (2) comparing the impact of different sampling strategies on hybrid graph learning methods; and (3) exploring ways to integrate simple graph and hypergraph information. We make our source code and full datasets publicly available at https://zehui127.github.io/hybrid-graph-benchmark/.
Toggling a Genetic Switch Using Reinforcement Learning
Feb 25, 2015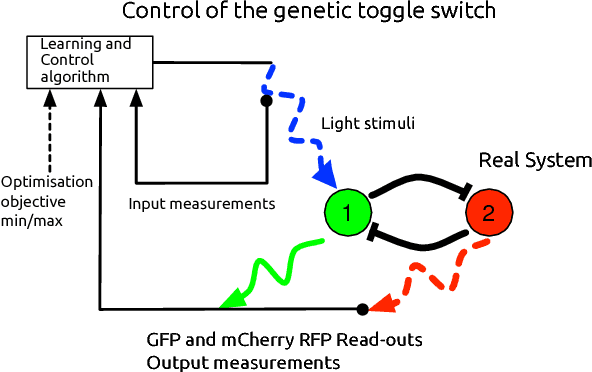
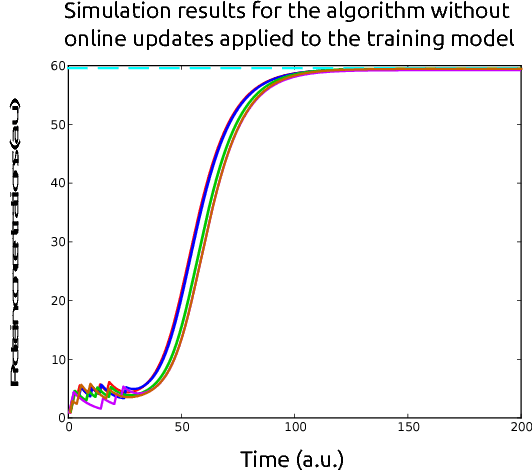
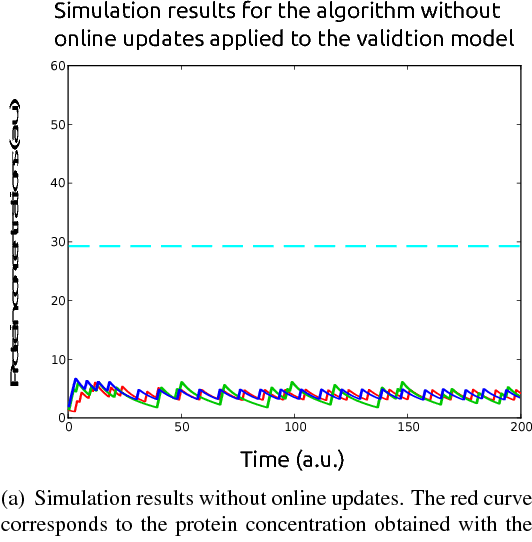
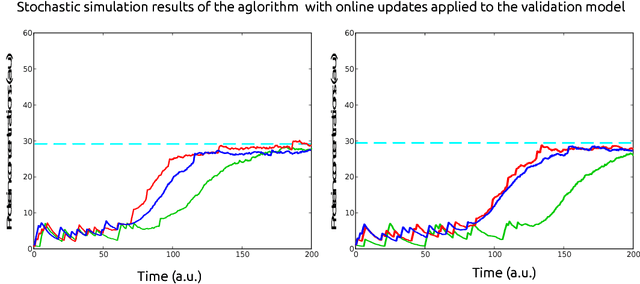
In this paper, we consider the problem of optimal exogenous control of gene regulatory networks. Our approach consists in adapting an established reinforcement learning algorithm called the fitted Q iteration. This algorithm infers the control law directly from the measurements of the system's response to external control inputs without the use of a mathematical model of the system. The measurement data set can either be collected from wet-lab experiments or artificially created by computer simulations of dynamical models of the system. The algorithm is applicable to a wide range of biological systems due to its ability to deal with nonlinear and stochastic system dynamics. To illustrate the application of the algorithm to a gene regulatory network, the regulation of the toggle switch system is considered. The control objective of this problem is to drive the concentrations of two specific proteins to a target region in the state space.
Distributed Reconstruction of Nonlinear Networks: An ADMM Approach
Mar 28, 2014
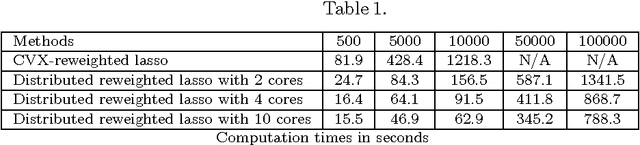
In this paper, we present a distributed algorithm for the reconstruction of large-scale nonlinear networks. In particular, we focus on the identification from time-series data of the nonlinear functional forms and associated parameters of large-scale nonlinear networks. Recently, a nonlinear network reconstruction problem was formulated as a nonconvex optimisation problem based on the combination of a marginal likelihood maximisation procedure with sparsity inducing priors. Using a convex-concave procedure (CCCP), an iterative reweighted lasso algorithm was derived to solve the initial nonconvex optimisation problem. By exploiting the structure of the objective function of this reweighted lasso algorithm, a distributed algorithm can be designed. To this end, we apply the alternating direction method of multipliers (ADMM) to decompose the original problem into several subproblems. To illustrate the effectiveness of the proposed methods, we use our approach to identify a network of interconnected Kuramoto oscillators with different network sizes (500~100,000 nodes).