 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAbsorb & Escape: Overcoming Single Model Limitations in Generating Genomic Sequences
Oct 28, 2024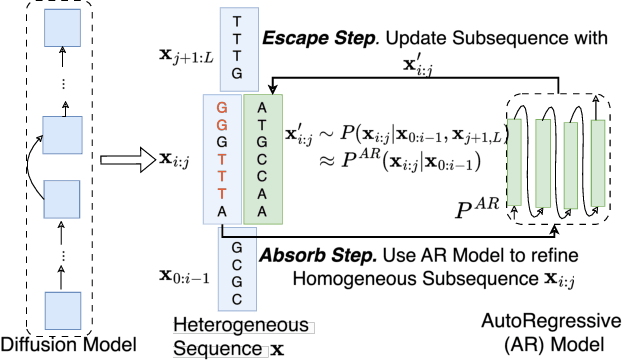


Abstract Recent advances in immunology and synthetic biology have accelerated the development of deep generative methods for DNA sequence design. Two dominant approaches in this field are AutoRegressive (AR) models and Diffusion Models (DMs). However, genomic sequences are functionally heterogeneous, consisting of multiple connected regions (e.g., Promoter Regions, Exons, and Introns) where elements within each region come from the same probability distribution, but the overall sequence is non-homogeneous. This heterogeneous nature presents challenges for a single model to accurately generate genomic sequences. In this paper, we analyze the properties of AR models and DMs in heterogeneous genomic sequence generation, pointing out crucial limitations in both methods: (i) AR models capture the underlying distribution of data by factorizing and learning the transition probability but fail to capture the global property of DNA sequences. (ii) DMs learn to recover the global distribution but tend to produce errors at the base pair level. To overcome the limitations of both approaches, we propose a post-training sampling method, termed Absorb & Escape (A&E) to perform compositional generation from AR models and DMs. This approach starts with samples generated by DMs and refines the sample quality using an AR model through the alternation of the Absorb and Escape steps. To assess the quality of generated sequences, we conduct extensive experiments on 15 species for conditional and unconditional DNA generation. The experiment results from motif distribution, diversity checks, and genome integration tests unequivocally show that A&E outperforms state-of-the-art AR models and DMs in genomic sequence generation.
DiscDiff: Latent Diffusion Model for DNA Sequence Generation
Feb 08, 2024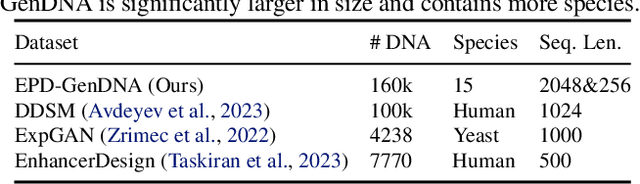
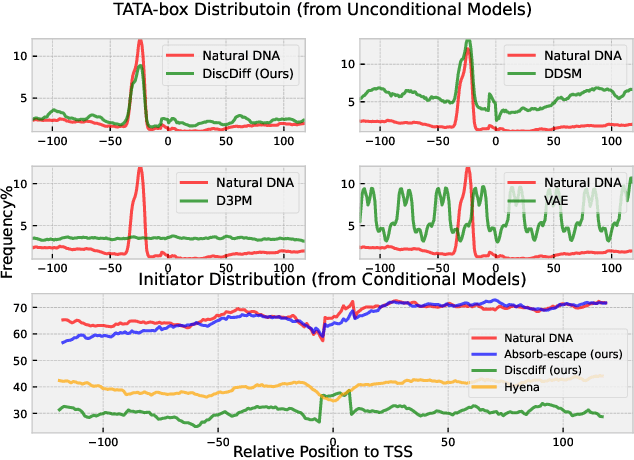

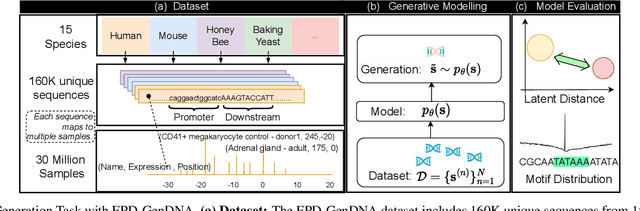
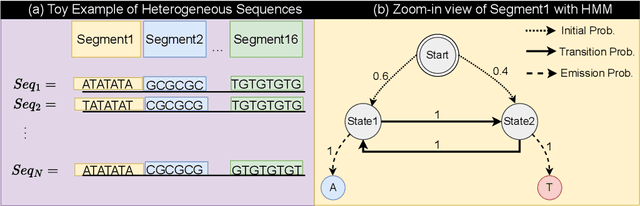
This paper introduces a novel framework for DNA sequence generation, comprising two key components: DiscDiff, a Latent Diffusion Model (LDM) tailored for generating discrete DNA sequences, and Absorb-Escape, a post-training algorithm designed to refine these sequences. Absorb-Escape enhances the realism of the generated sequences by correcting `round errors' inherent in the conversion process between latent and input spaces. Our approach not only sets new standards in DNA sequence generation but also demonstrates superior performance over existing diffusion models, in generating both short and long DNA sequences. Additionally, we introduce EPD-GenDNA, the first comprehensive, multi-species dataset for DNA generation, encompassing 160,000 unique sequences from 15 species. We hope this study will advance the generative modelling of DNA, with potential implications for gene therapy and protein production.
Latent Diffusion Model for DNA Sequence Generation
Oct 09, 2023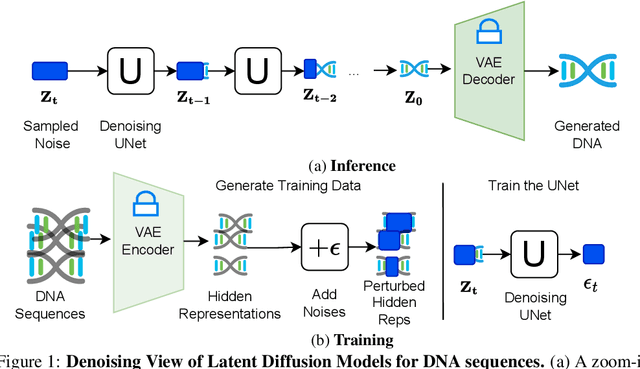
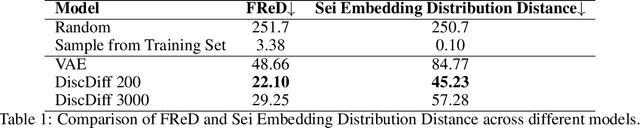
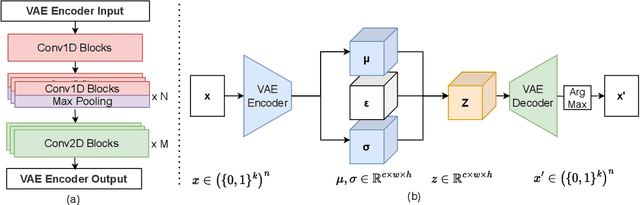
The harnessing of machine learning, especially deep generative models, has opened up promising avenues in the field of synthetic DNA sequence generation. Whilst Generative Adversarial Networks (GANs) have gained traction for this application, they often face issues such as limited sample diversity and mode collapse. On the other hand, Diffusion Models are a promising new class of generative models that are not burdened with these problems, enabling them to reach the state-of-the-art in domains such as image generation. In light of this, we propose a novel latent diffusion model, DiscDiff, tailored for discrete DNA sequence generation. By simply embedding discrete DNA sequences into a continuous latent space using an autoencoder, we are able to leverage the powerful generative abilities of continuous diffusion models for the generation of discrete data. Additionally, we introduce Fr\'echet Reconstruction Distance (FReD) as a new metric to measure the sample quality of DNA sequence generations. Our DiscDiff model demonstrates an ability to generate synthetic DNA sequences that align closely with real DNA in terms of Motif Distribution, Latent Embedding Distribution (FReD), and Chromatin Profiles. Additionally, we contribute a comprehensive cross-species dataset of 150K unique promoter-gene sequences from 15 species, enriching resources for future generative modelling in genomics. We will make our code public upon publication.
Genomic Interpreter: A Hierarchical Genomic Deep Neural Network with 1D Shifted Window Transformer
Jun 28, 2023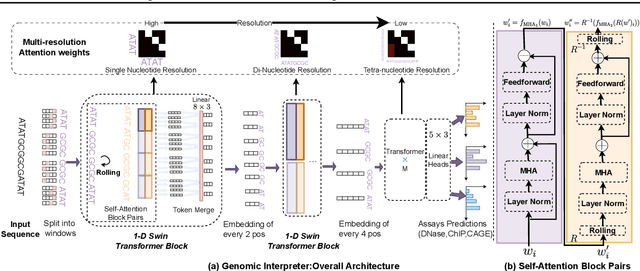
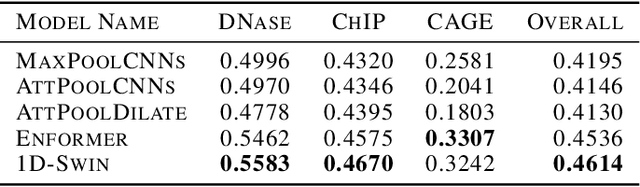
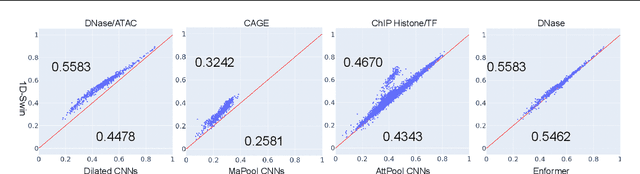
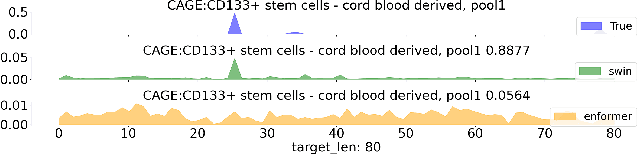
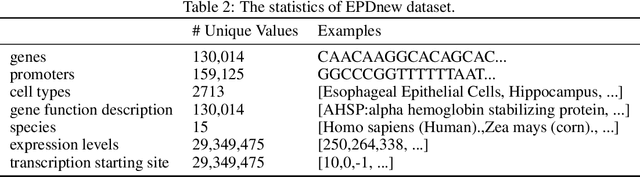
Given the increasing volume and quality of genomics data, extracting new insights requires interpretable machine-learning models. This work presents Genomic Interpreter: a novel architecture for genomic assay prediction. This model outperforms the state-of-the-art models for genomic assay prediction tasks. Our model can identify hierarchical dependencies in genomic sites. This is achieved through the integration of 1D-Swin, a novel Transformer-based block designed by us for modelling long-range hierarchical data. Evaluated on a dataset containing 38,171 DNA segments of 17K base pairs, Genomic Interpreter demonstrates superior performance in chromatin accessibility and gene expression prediction and unmasks the underlying `syntax' of gene regulation.