 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApo2Mol: 3D Molecule Generation via Dynamic Pocket-Aware Diffusion Models
Nov 18, 2025



Deep generative models are rapidly advancing structure-based drug design, offering substantial promise for generating small molecule ligands that bind to specific protein targets. However, most current approaches assume a rigid protein binding pocket, neglecting the intrinsic flexibility of proteins and the conformational rearrangements induced by ligand binding, limiting their applicability in practical drug discovery. Here, we propose Apo2Mol, a diffusion-based generative framework for 3D molecule design that explicitly accounts for conformational flexibility in protein binding pockets. To support this, we curate a dataset of over 24,000 experimentally resolved apo-holo structure pairs from the Protein Data Bank, enabling the characterization of protein structure changes associated with ligand binding. Apo2Mol employs a full-atom hierarchical graph-based diffusion model that simultaneously generates 3D ligand molecules and their corresponding holo pocket conformations from input apo states. Empirical studies demonstrate that Apo2Mol can achieve state-of-the-art performance in generating high-affinity ligands and accurately capture realistic protein pocket conformational changes.
Large Language Model for Extracting Complex Contract Information in Industrial Scenes
Jul 09, 2025



This paper proposes a high-quality dataset construction method for complex contract information extraction tasks in industrial scenarios and fine-tunes a large language model based on this dataset. Firstly, cluster analysis is performed on industrial contract texts, and GPT-4 and GPT-3.5 are used to extract key information from the original contract data, obtaining high-quality data annotations. Secondly, data augmentation is achieved by constructing new texts, and GPT-3.5 generates unstructured contract texts from randomly combined keywords, improving model robustness. Finally, the large language model is fine-tuned based on the high-quality dataset. Experimental results show that the model achieves excellent overall performance while ensuring high field recall and precision and considering parsing efficiency. LoRA, data balancing, and data augmentation effectively enhance model accuracy and robustness. The proposed method provides a novel and efficient solution for industrial contract information extraction tasks.
DecoyDB: A Dataset for Graph Contrastive Learning in Protein-Ligand Binding Affinity Prediction
Jul 08, 2025Predicting the binding affinity of protein-ligand complexes plays a vital role in drug discovery. Unfortunately, progress has been hindered by the lack of large-scale and high-quality binding affinity labels. The widely used PDBbind dataset has fewer than 20K labeled complexes. Self-supervised learning, especially graph contrastive learning (GCL), provides a unique opportunity to break the barrier by pre-training graph neural network models based on vast unlabeled complexes and fine-tuning the models on much fewer labeled complexes. However, the problem faces unique challenges, including a lack of a comprehensive unlabeled dataset with well-defined positive/negative complex pairs and the need to design GCL algorithms that incorporate the unique characteristics of such data. To fill the gap, we propose DecoyDB, a large-scale, structure-aware dataset specifically designed for self-supervised GCL on protein-ligand complexes. DecoyDB consists of high-resolution ground truth complexes (less than 2.5 Angstrom) and diverse decoy structures with computationally generated binding poses that range from realistic to suboptimal (negative pairs). Each decoy is annotated with a Root Mean Squared Deviation (RMSD) from the native pose. We further design a customized GCL framework to pre-train graph neural networks based on DecoyDB and fine-tune the models with labels from PDBbind. Extensive experiments confirm that models pre-trained with DecoyDB achieve superior accuracy, label efficiency, and generalizability.
HSACNet: Hierarchical Scale-Aware Consistency Regularized Semi-Supervised Change Detection
Apr 18, 2025Semi-supervised change detection (SSCD) aims to detect changes between bi-temporal remote sensing images by utilizing limited labeled data and abundant unlabeled data. Existing methods struggle in complex scenarios, exhibiting poor performance when confronted with noisy data. They typically neglect intra-layer multi-scale features while emphasizing inter-layer fusion, harming the integrity of change objects with different scales. In this paper, we propose HSACNet, a Hierarchical Scale-Aware Consistency regularized Network for SSCD. Specifically, we integrate Segment Anything Model 2 (SAM2), using its Hiera backbone as the encoder to extract inter-layer multi-scale features and applying adapters for parameter-efficient fine-tuning. Moreover, we design a Scale-Aware Differential Attention Module (SADAM) that can precisely capture intra-layer multi-scale change features and suppress noise. Additionally, a dual-augmentation consistency regularization strategy is adopted to effectively utilize the unlabeled data. Extensive experiments across four CD benchmarks demonstrate that our HSACNet achieves state-of-the-art performance, with reduced parameters and computational cost.
SegBook: A Simple Baseline and Cookbook for Volumetric Medical Image Segmentation
Nov 21, 2024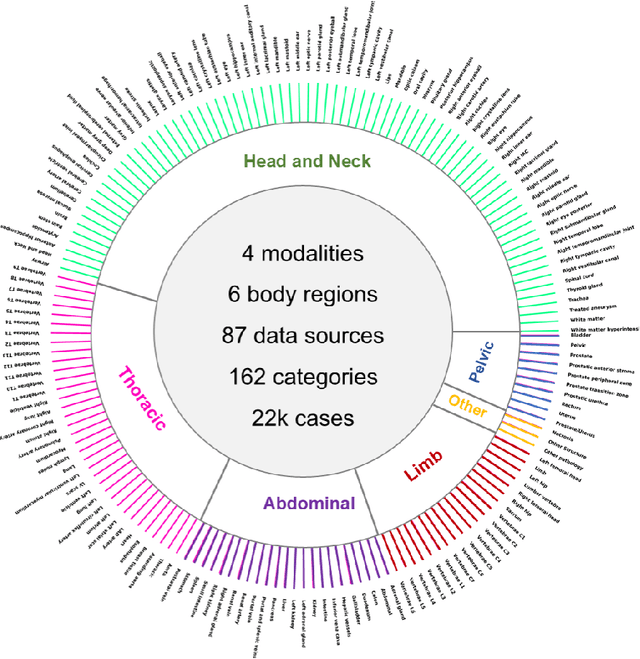
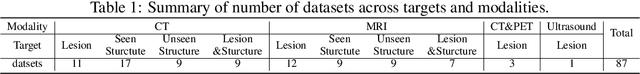
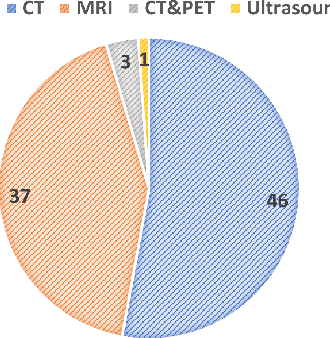
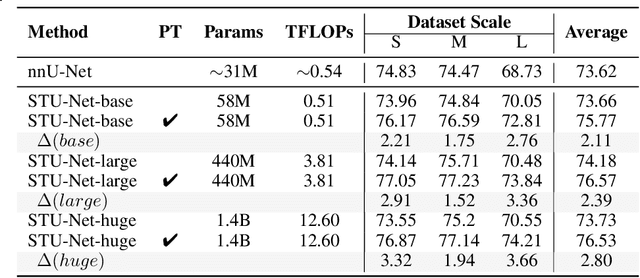
Computed Tomography (CT) is one of the most popular modalities for medical imaging. By far, CT images have contributed to the largest publicly available datasets for volumetric medical segmentation tasks, covering full-body anatomical structures. Large amounts of full-body CT images provide the opportunity to pre-train powerful models, e.g., STU-Net pre-trained in a supervised fashion, to segment numerous anatomical structures. However, it remains unclear in which conditions these pre-trained models can be transferred to various downstream medical segmentation tasks, particularly segmenting the other modalities and diverse targets. To address this problem, a large-scale benchmark for comprehensive evaluation is crucial for finding these conditions. Thus, we collected 87 public datasets varying in modality, target, and sample size to evaluate the transfer ability of full-body CT pre-trained models. We then employed a representative model, STU-Net with multiple model scales, to conduct transfer learning across modalities and targets. Our experimental results show that (1) there may be a bottleneck effect concerning the dataset size in fine-tuning, with more improvement on both small- and large-scale datasets than medium-size ones. (2) Models pre-trained on full-body CT demonstrate effective modality transfer, adapting well to other modalities such as MRI. (3) Pre-training on the full-body CT not only supports strong performance in structure detection but also shows efficacy in lesion detection, showcasing adaptability across target tasks. We hope that this large-scale open evaluation of transfer learning can direct future research in volumetric medical image segmentation.
GMAI-VL & GMAI-VL-5.5M: A Large Vision-Language Model and A Comprehensive Multimodal Dataset Towards General Medical AI
Nov 21, 2024Despite significant advancements in general artificial intelligence, such as GPT-4, their effectiveness in the medical domain (general medical AI, GMAI) remains constrained due to the absence of specialized medical knowledge. To address this challenge, we present GMAI-VL-5.5M, a comprehensive multimodal medical dataset created by converting hundreds of specialized medical datasets into meticulously constructed image-text pairs. This dataset features comprehensive task coverage, diverse modalities, and high-quality image-text data. Building upon this multimodal dataset, we propose GMAI-VL, a general medical vision-language model with a progressively three-stage training strategy. This approach significantly enhances the model's ability by integrating visual and textual information, thereby improving its ability to process multimodal data and support accurate diagnosis and clinical decision-making. Experimental evaluations demonstrate that GMAI-VL achieves state-of-the-art results across a wide range of multimodal medical tasks, such as visual question answering and medical image diagnosis. Our contributions include the development of the GMAI-VL-5.5M dataset, the introduction of the GMAI-VL model, and the establishment of new benchmarks in multiple medical domains. Code and dataset will be released at https://github.com/uni-medical/GMAI-VL.
SlideChat: A Large Vision-Language Assistant for Whole-Slide Pathology Image Understanding
Oct 15, 2024



Despite the progress made by multimodal large language models (MLLMs) in computational pathology, they remain limited by a predominant focus on patch-level analysis, missing essential contextual information at the whole-slide level. The lack of large-scale instruction datasets and the gigapixel scale of whole slide images (WSIs) pose significant developmental challenges. In this paper, we present SlideChat, the first vision-language assistant capable of understanding gigapixel whole-slide images, exhibiting excellent multimodal conversational capability and response complex instruction across diverse pathology scenarios. To support its development, we created SlideInstruction, the largest instruction-following dataset for WSIs consisting of 4.2K WSI captions and 176K VQA pairs with multiple categories. Furthermore, we propose SlideBench, a multimodal benchmark that incorporates captioning and VQA tasks to assess SlideChat's capabilities in varied clinical settings such as microscopy, diagnosis. Compared to both general and specialized MLLMs, SlideChat exhibits exceptional capabilities achieving state-of-the-art performance on 18 of 22 tasks. For example, it achieved an overall accuracy of 81.17% on SlideBench-VQA (TCGA), and 54.15% on SlideBench-VQA (BCNB). We will fully release SlideChat, SlideInstruction and SlideBench as open-source resources to facilitate research and development in computational pathology.
GMAI-MMBench: A Comprehensive Multimodal Evaluation Benchmark Towards General Medical AI
Aug 06, 2024Large Vision-Language Models (LVLMs) are capable of handling diverse data types such as imaging, text, and physiological signals, and can be applied in various fields. In the medical field, LVLMs have a high potential to offer substantial assistance for diagnosis and treatment. Before that, it is crucial to develop benchmarks to evaluate LVLMs' effectiveness in various medical applications. Current benchmarks are often built upon specific academic literature, mainly focusing on a single domain, and lacking varying perceptual granularities. Thus, they face specific challenges, including limited clinical relevance, incomplete evaluations, and insufficient guidance for interactive LVLMs. To address these limitations, we developed the GMAI-MMBench, the most comprehensive general medical AI benchmark with well-categorized data structure and multi-perceptual granularity to date. It is constructed from 285 datasets across 39 medical image modalities, 18 clinical-related tasks, 18 departments, and 4 perceptual granularities in a Visual Question Answering (VQA) format. Additionally, we implemented a lexical tree structure that allows users to customize evaluation tasks, accommodating various assessment needs and substantially supporting medical AI research and applications. We evaluated 50 LVLMs, and the results show that even the advanced GPT-4o only achieves an accuracy of 52\%, indicating significant room for improvement. Moreover, we identified five key insufficiencies in current cutting-edge LVLMs that need to be addressed to advance the development of better medical applications. We believe that GMAI-MMBench will stimulate the community to build the next generation of LVLMs toward GMAI.
Morphological Profiling for Drug Discovery in the Era of Deep Learning
Dec 13, 2023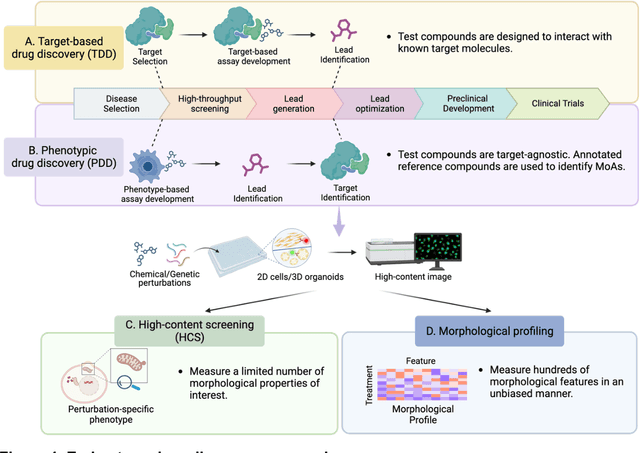
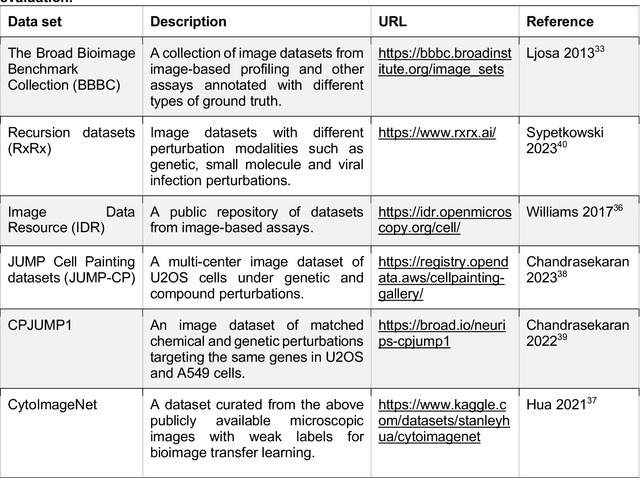
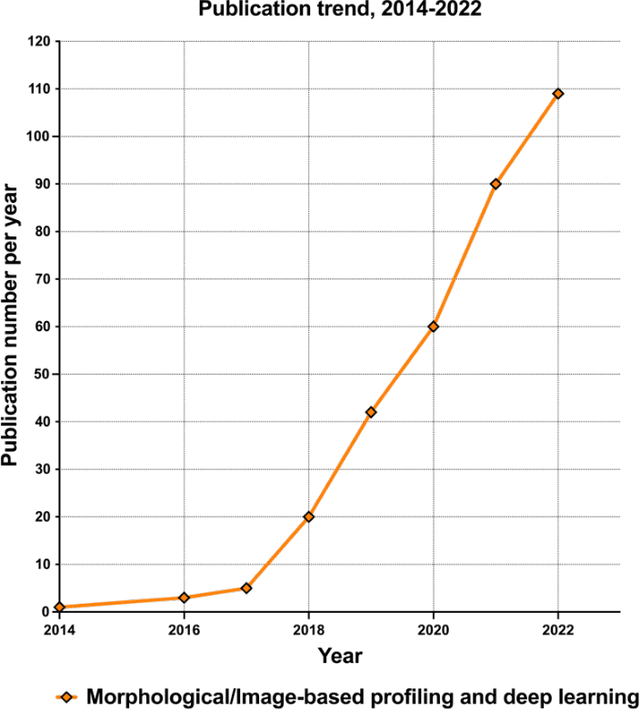

Morphological profiling is a valuable tool in phenotypic drug discovery. The advent of high-throughput automated imaging has enabled the capturing of a wide range of morphological features of cells or organisms in response to perturbations at the single-cell resolution. Concurrently, significant advances in machine learning and deep learning, especially in computer vision, have led to substantial improvements in analyzing large-scale high-content images at high-throughput. These efforts have facilitated understanding of compound mechanism-of-action (MOA), drug repurposing, characterization of cell morphodynamics under perturbation, and ultimately contributing to the development of novel therapeutics. In this review, we provide a comprehensive overview of the recent advances in the field of morphological profiling. We summarize the image profiling analysis workflow, survey a broad spectrum of analysis strategies encompassing feature engineering- and deep learning-based approaches, and introduce publicly available benchmark datasets. We place a particular emphasis on the application of deep learning in this pipeline, covering cell segmentation, image representation learning, and multimodal learning. Additionally, we illuminate the application of morphological profiling in phenotypic drug discovery and highlight potential challenges and opportunities in this field.
Identifying acute illness phenotypes via deep temporal interpolation and clustering network on physiologic signatures
Jul 27, 2023Initial hours of hospital admission impact clinical trajectory, but early clinical decisions often suffer due to data paucity. With clustering analysis for vital signs within six hours of admission, patient phenotypes with distinct pathophysiological signatures and outcomes may support early clinical decisions. We created a single-center, longitudinal EHR dataset for 75,762 adults admitted to a tertiary care center for 6+ hours. We proposed a deep temporal interpolation and clustering network to extract latent representations from sparse, irregularly sampled vital sign data and derived distinct patient phenotypes in a training cohort (n=41,502). Model and hyper-parameters were chosen based on a validation cohort (n=17,415). Test cohort (n=16,845) was used to analyze reproducibility and correlation with biomarkers. The training, validation, and testing cohorts had similar distributions of age (54-55 yrs), sex (55% female), race, comorbidities, and illness severity. Four clusters were identified. Phenotype A (18%) had most comorbid disease with higher rate of prolonged respiratory insufficiency, acute kidney injury, sepsis, and three-year mortality. Phenotypes B (33%) and C (31%) had diffuse patterns of mild organ dysfunction. Phenotype B had favorable short-term outcomes but second-highest three-year mortality. Phenotype C had favorable clinical outcomes. Phenotype D (17%) had early/persistent hypotension, high rate of early surgery, and substantial biomarker rate of inflammation but second-lowest three-year mortality. After comparing phenotypes' SOFA scores, clustering results did not simply repeat other acuity assessments. In a heterogeneous cohort, four phenotypes with distinct categories of disease and outcomes were identified by a deep temporal interpolation and clustering network. This tool may impact triage decisions and clinical decision-support under time constraints.