 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDrVD-Bench: Do Vision-Language Models Reason Like Human Doctors in Medical Image Diagnosis?
May 30, 2025Vision-language models (VLMs) exhibit strong zero-shot generalization on natural images and show early promise in interpretable medical image analysis. However, existing benchmarks do not systematically evaluate whether these models truly reason like human clinicians or merely imitate superficial patterns. To address this gap, we propose DrVD-Bench, the first multimodal benchmark for clinical visual reasoning. DrVD-Bench consists of three modules: Visual Evidence Comprehension, Reasoning Trajectory Assessment, and Report Generation Evaluation, comprising a total of 7,789 image-question pairs. Our benchmark covers 20 task types, 17 diagnostic categories, and five imaging modalities-CT, MRI, ultrasound, radiography, and pathology. DrVD-Bench is explicitly structured to reflect the clinical reasoning workflow from modality recognition to lesion identification and diagnosis. We benchmark 19 VLMs, including general-purpose and medical-specific, open-source and proprietary models, and observe that performance drops sharply as reasoning complexity increases. While some models begin to exhibit traces of human-like reasoning, they often still rely on shortcut correlations rather than grounded visual understanding. DrVD-Bench offers a rigorous and structured evaluation framework to guide the development of clinically trustworthy VLMs.
Benchmarking AI scientists in omics data-driven biological research
May 13, 2025The rise of large language models and multi-agent systems has sparked growing interest in AI scientists capable of autonomous biological research. However, existing benchmarks either focus on reasoning without data or on data analysis with predefined statistical answers, lacking realistic, data-driven evaluation settings. Here, we introduce the Biological AI Scientist Benchmark (BaisBench), a benchmark designed to assess AI scientists' ability to generate biological discoveries through data analysis and reasoning with external knowledge. BaisBench comprises two tasks: cell type annotation on 31 expert-labeled single-cell datasets, and scientific discovery through answering 198 multiple-choice questions derived from the biological insights of 41 recent single-cell studies. Systematic experiments on state-of-the-art AI scientists and LLM agents showed that while promising, current models still substantially underperform human experts on both tasks. We hope BaisBench will fill this gap and serve as a foundation for advancing and evaluating AI models for scientific discovery. The benchmark can be found at: https://github.com/EperLuo/BaisBench.
QueST: Querying Functional and Structural Niches on Spatial Transcriptomics Data via Contrastive Subgraph Embedding
Oct 14, 2024



The functional or structural spatial regions within tissues, referred to as spatial niches, are elements for illustrating the spatial contexts of multicellular organisms. A key challenge is querying shared niches across diverse tissues, which is crucial for achieving a comprehensive understanding of the organization and phenotypes of cell populations. However, current data analysis methods predominantly focus on creating spatial-aware embeddings for cells, neglecting the development of niche-level representations for effective querying. To address this gap, we introduce QueST, a novel niche representation learning model designed for querying spatial niches across multiple samples. QueST utilizes a novel subgraph contrastive learning approach to explicitly capture niche-level characteristics and incorporates adversarial training to mitigate batch effects. We evaluate QueST on established benchmarks using human and mouse datasets, demonstrating its superiority over state-of-the-art graph representation learning methods in accurate niche queries. Overall, QueST offers a specialized model for spatial niche queries, paving the way for deeper insights into the patterns and mechanisms of cell spatial organization across tissues. Source code can be found at https://github.com/cmhimself/QueST.
Molecular Graph Representation Learning Integrating Large Language Models with Domain-specific Small Models
Aug 19, 2024



Molecular property prediction is a crucial foundation for drug discovery. In recent years, pre-trained deep learning models have been widely applied to this task. Some approaches that incorporate prior biological domain knowledge into the pre-training framework have achieved impressive results. However, these methods heavily rely on biochemical experts, and retrieving and summarizing vast amounts of domain knowledge literature is both time-consuming and expensive. Large Language Models (LLMs) have demonstrated remarkable performance in understanding and efficiently providing general knowledge. Nevertheless, they occasionally exhibit hallucinations and lack precision in generating domain-specific knowledge. Conversely, Domain-specific Small Models (DSMs) possess rich domain knowledge and can accurately calculate molecular domain-related metrics. However, due to their limited model size and singular functionality, they lack the breadth of knowledge necessary for comprehensive representation learning. To leverage the advantages of both approaches in molecular property prediction, we propose a novel Molecular Graph representation learning framework that integrates Large language models and Domain-specific small models (MolGraph-LarDo). Technically, we design a two-stage prompt strategy where DSMs are introduced to calibrate the knowledge provided by LLMs, enhancing the accuracy of domain-specific information and thus enabling LLMs to generate more precise textual descriptions for molecular samples. Subsequently, we employ a multi-modal alignment method to coordinate various modalities, including molecular graphs and their corresponding descriptive texts, to guide the pre-training of molecular representations. Extensive experiments demonstrate the effectiveness of the proposed method.
Weakly-supervised causal discovery based on fuzzy knowledge and complex data complementarity
May 14, 2024Causal discovery based on observational data is important for deciphering the causal mechanism behind complex systems. However, the effectiveness of existing causal discovery methods is limited due to inferior prior knowledge, domain inconsistencies, and the challenges of high-dimensional datasets with small sample sizes. To address this gap, we propose a novel weakly-supervised fuzzy knowledge and data co-driven causal discovery method named KEEL. KEEL adopts a fuzzy causal knowledge schema to encapsulate diverse types of fuzzy knowledge, and forms corresponding weakened constraints. This schema not only lessens the dependency on expertise but also allows various types of limited and error-prone fuzzy knowledge to guide causal discovery. It can enhance the generalization and robustness of causal discovery, especially in high-dimensional and small-sample scenarios. In addition, we integrate the extended linear causal model (ELCM) into KEEL for dealing with the multi-distribution and incomplete data. Extensive experiments with different datasets demonstrate the superiority of KEEL over several state-of-the-art methods in accuracy, robustness and computational efficiency. For causal discovery in real protein signal transduction processes, KEEL outperforms the benchmark method with limited data. In summary, KEEL is effective to tackle the causal discovery tasks with higher accuracy while alleviating the requirement for extensive domain expertise.
Label Informed Contrastive Pretraining for Node Importance Estimation on Knowledge Graphs
Feb 26, 2024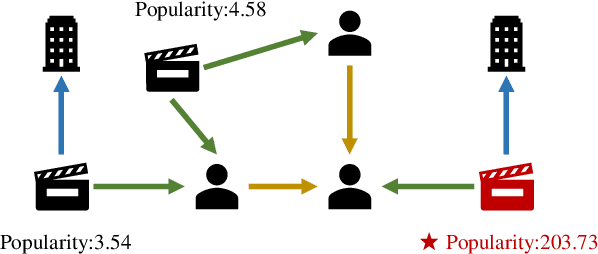
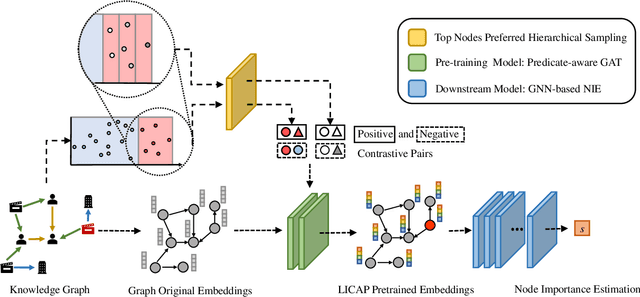
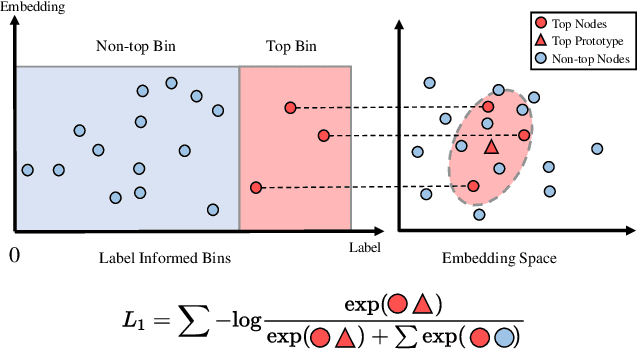
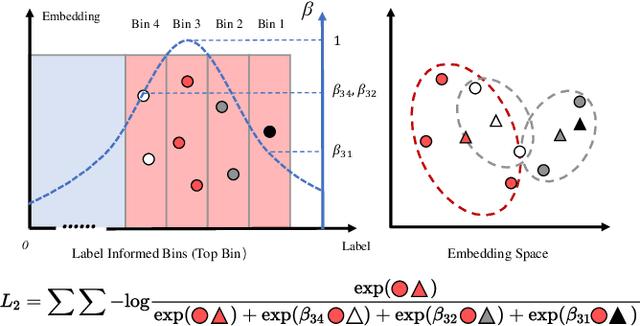
Node Importance Estimation (NIE) is a task of inferring importance scores of the nodes in a graph. Due to the availability of richer data and knowledge, recent research interests of NIE have been dedicating to knowledge graphs for predicting future or missing node importance scores. Existing state-of-the-art NIE methods train the model by available labels, and they consider every interested node equally before training. However, the nodes with higher importance often require or receive more attention in real-world scenarios, e.g., people may care more about the movies or webpages with higher importance. To this end, we introduce Label Informed ContrAstive Pretraining (LICAP) to the NIE problem for being better aware of the nodes with high importance scores. Specifically, LICAP is a novel type of contrastive learning framework that aims to fully utilize the continuous labels to generate contrastive samples for pretraining embeddings. Considering the NIE problem, LICAP adopts a novel sampling strategy called top nodes preferred hierarchical sampling to first group all interested nodes into a top bin and a non-top bin based on node importance scores, and then divide the nodes within top bin into several finer bins also based on the scores. The contrastive samples are generated from those bins, and are then used to pretrain node embeddings of knowledge graphs via a newly proposed Predicate-aware Graph Attention Networks (PreGAT), so as to better separate the top nodes from non-top nodes, and distinguish the top nodes within top bin by keeping the relative order among finer bins. Extensive experiments demonstrate that the LICAP pretrained embeddings can further boost the performance of existing NIE methods and achieve the new state-of-the-art performance regarding both regression and ranking metrics. The source code for reproducibility is available at https://github.com/zhangtia16/LICAP
scDiffusion: conditional generation of high-quality single-cell data using diffusion model
Jan 08, 2024



Single-cell RNA sequencing (scRNA-seq) data are important for studying the biology of development or diseases at single-cell level. To better understand the properties of the data, to build controlled benchmark data for testing downstream methods, and to augment data when collecting sufficient real data is challenging, generative models have been proposed to computationally generate synthetic scRNA-seq data. However, the data generated with current models are not very realistic yet, especially when we need to generate data with controlled conditions. In the meantime, the Diffusion models have shown their power in generating data in computer vision at high fidelity, providing a new opportunity for scRNA-seq generation. In this study, we developed scDiffusion, a diffusion-based model to generate high-quality scRNA-seq data with controlled conditions. We designed multiple classifiers to guide the diffusion process simultaneously, enabling scDiffusion to generate data under multiple condition combinations. We also proposed a new control strategy called Gradient Interpolation. This strategy allows the model to generate continuous trajectories of cell development from a given cell state. Experiments showed that scDiffusion can generate single-cell gene expression data closely resembling real scRNA-seq data, surpassing state-of-the-art models in multiple metrics. Also, scDiffusion can conditionally produce data on specific cell types including rare cell types. Furthermore, we could use the multiple-condition generation of scDiffusion to generate cell type that was out of the training data. Leveraging the Gradient Interpolation strategy, we generated a continuous developmental trajectory of mouse embryonic cells. These experiments demonstrate that scDiffusion is a powerful tool for augmenting the real scRNA-seq data and can provide insights into cell fate research.
xTrimoGene: An Efficient and Scalable Representation Learner for Single-Cell RNA-Seq Data
Nov 26, 2023Advances in high-throughput sequencing technology have led to significant progress in measuring gene expressions at the single-cell level. The amount of publicly available single-cell RNA-seq (scRNA-seq) data is already surpassing 50M records for humans with each record measuring 20,000 genes. This highlights the need for unsupervised representation learning to fully ingest these data, yet classical transformer architectures are prohibitive to train on such data in terms of both computation and memory. To address this challenge, we propose a novel asymmetric encoder-decoder transformer for scRNA-seq data, called xTrimoGene$^\alpha$ (or xTrimoGene for short), which leverages the sparse characteristic of the data to scale up the pre-training. This scalable design of xTrimoGene reduces FLOPs by one to two orders of magnitude compared to classical transformers while maintaining high accuracy, enabling us to train the largest transformer models over the largest scRNA-seq dataset today. Our experiments also show that the performance of xTrimoGene improves as we scale up the model sizes, and it also leads to SOTA performance over various downstream tasks, such as cell type annotation, perturb-seq effect prediction, and drug combination prediction. xTrimoGene model is now available for use as a service via the following link: https://api.biomap.com/xTrimoGene/apply.
MMA-RNN: A Multi-level Multi-task Attention-based Recurrent Neural Network for Discrimination and Localization of Atrial Fibrillation
Feb 09, 2023The automatic detection of atrial fibrillation based on electrocardiograph (ECG) signals has received wide attention both clinically and practically. It is challenging to process ECG signals with cyclical pattern, varying length and unstable quality due to noise and distortion. Besides, there has been insufficient research on separating persistent atrial fibrillation from paroxysmal atrial fibrillation, and little discussion on locating the onsets and end points of AF episodes. It is even more arduous to perform well on these two distinct but interrelated tasks, while avoiding the mistakes inherent from stage-by-stage approaches. This paper proposes the Multi-level Multi-task Attention-based Recurrent Neural Network for three-class discrimination on patients and localization of the exact timing of AF episodes. Our model captures three-level sequential features based on a hierarchical architecture utilizing Bidirectional Long and Short-Term Memory Network (Bi-LSTM) and attention layers, and accomplishes the two tasks simultaneously with a multi-head classifier. The model is designed as an end-to-end framework to enhance information interaction and reduce error accumulation. Finally, we conduct experiments on CPSC 2021 dataset and the result demonstrates the superior performance of our method, indicating the potential application of MMA-RNN to wearable mobile devices for routine AF monitoring and early diagnosis.
Generation of Synthetic Electronic Medical Record Text
Dec 06, 2018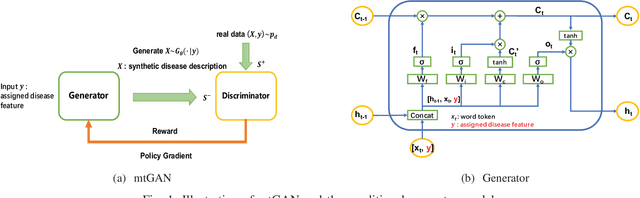
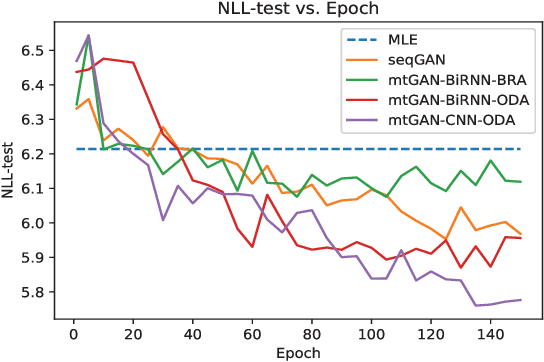


Machine learning (ML) and Natural Language Processing (NLP) have achieved remarkable success in many fields and have brought new opportunities and high expectation in the analyses of medical data. The most common type of medical data is the massive free-text electronic medical records (EMR). It is widely regarded that mining such massive data can bring up important information for improving medical practices as well as for possible new discoveries on complex diseases. However, the free EMR texts are lacking consistent standards, rich of private information, and limited in availability. Also, as they are accumulated from everyday practices, it is often hard to have a balanced number of samples for the types of diseases under study. These problems hinder the development of ML and NLP methods for EMR data analysis. To tackle these problems, we developed a model to generate synthetic text of EMRs called Medical Text Generative Adversarial Network or mtGAN. It is based on the GAN framework and is trained by the REINFORCE algorithm. It takes disease features as inputs and generates synthetic texts as EMRs for the corresponding diseases. We evaluate the model from micro-level, macro-level and application-level on a Chinese EMR text dataset. The results show that the method has a good capacity to fit real data and can generate realistic and diverse EMR samples. This provides a novel way to avoid potential leakage of patient privacy while still supply sufficient well-controlled cohort data for developing downstream ML and NLP methods. It can also be used as a data augmentation method to assist studies based on real EMR data.