 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRAGRouter-Bench: A Dataset and Benchmark for Adaptive RAG Routing
Jan 30, 2026Retrieval-Augmented Generation (RAG) has become a core paradigm for grounding large language models with external knowledge. Despite extensive efforts exploring diverse retrieval strategies, existing studies predominantly focus on query-side complexity or isolated method improvements, lacking a systematic understanding of how RAG paradigms behave across different query-corpus contexts and effectiveness-efficiency trade-offs. In this work, we introduce RAGRouter-Bench, the first dataset and benchmark designed for adaptive RAG routing. RAGRouter-Bench revisits retrieval from a query-corpus compatibility perspective and standardizes five representative RAG paradigms for systematic evaluation across 7,727 queries and 21,460 documents spanning diverse domains. The benchmark incorporates three canonical query types together with fine-grained semantic and structural corpus metrics, as well as a unified evaluation for both generation quality and resource consumption. Experiments with DeepSeek-V3 and LLaMA-3.1-8B demonstrate that no single RAG paradigm is universally optimal, that paradigm applicability is strongly shaped by query-corpus interactions, and that increased advanced mechanism does not necessarily yield better effectiveness-efficiency trade-offs. These findings underscore the necessity of routing-aware evaluation and establish a foundation for adaptive, interpretable, and generalizable next-generation RAG systems.
GLaD: Geometric Latent Distillation for Vision-Language-Action Models
Dec 10, 2025
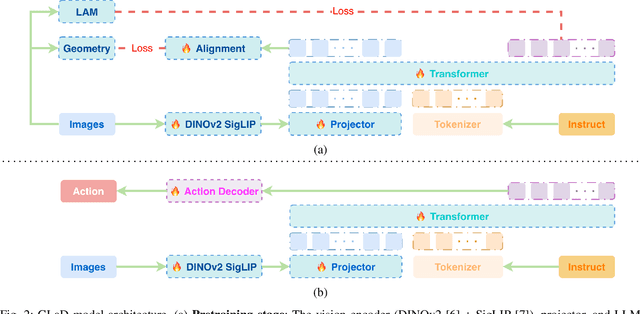
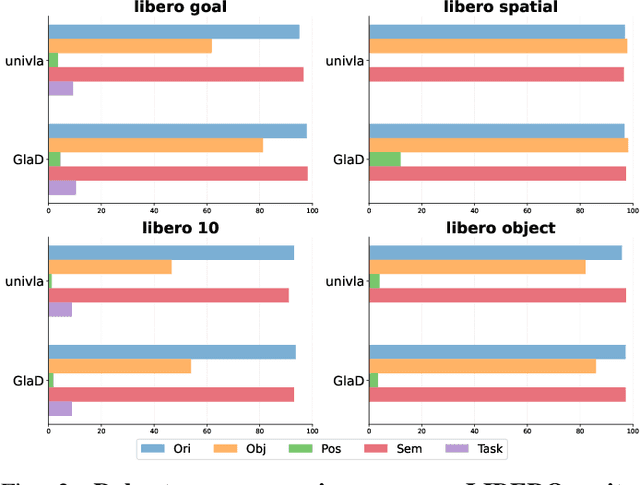

Most existing Vision-Language-Action (VLA) models rely primarily on RGB information, while ignoring geometric cues crucial for spatial reasoning and manipulation. In this work, we introduce GLaD, a geometry-aware VLA framework that incorporates 3D geometric priors during pretraining through knowledge distillation. Rather than distilling geometric features solely into the vision encoder, we align the LLM's hidden states corresponding to visual tokens with features from a frozen geometry-aware vision transformer (VGGT), ensuring that geometric understanding is deeply integrated into the multimodal representations that drive action prediction. Pretrained on the Bridge dataset with this geometry distillation mechanism, GLaD achieves 94.1% average success rate across four LIBERO task suites, outperforming UniVLA (92.5%) which uses identical pretraining data. These results validate that geometry-aware pretraining enhances spatial reasoning and policy generalization without requiring explicit depth sensors or 3D annotations.
Leveraging Geometric Visual Illusions as Perceptual Inductive Biases for Vision Models
Sep 18, 2025Contemporary deep learning models have achieved impressive performance in image classification by primarily leveraging statistical regularities within large datasets, but they rarely incorporate structured insights drawn directly from perceptual psychology. To explore the potential of perceptually motivated inductive biases, we propose integrating classic geometric visual illusions well-studied phenomena from human perception into standard image-classification training pipelines. Specifically, we introduce a synthetic, parametric geometric-illusion dataset and evaluate three multi-source learning strategies that combine illusion recognition tasks with ImageNet classification objectives. Our experiments reveal two key conceptual insights: (i) incorporating geometric illusions as auxiliary supervision systematically improves generalization, especially in visually challenging cases involving intricate contours and fine textures; and (ii) perceptually driven inductive biases, even when derived from synthetic stimuli traditionally considered unrelated to natural image recognition, can enhance the structural sensitivity of both CNN and transformer-based architectures. These results demonstrate a novel integration of perceptual science and machine learning and suggest new directions for embedding perceptual priors into vision model design.
DeepSieve: Information Sieving via LLM-as-a-Knowledge-Router
Jul 30, 2025Large Language Models (LLMs) excel at many reasoning tasks but struggle with knowledge-intensive queries due to their inability to dynamically access up-to-date or domain-specific information. Retrieval-Augmented Generation (RAG) has emerged as a promising solution, enabling LLMs to ground their responses in external sources. However, existing RAG methods lack fine-grained control over both the query and source sides, often resulting in noisy retrieval and shallow reasoning. In this work, we introduce DeepSieve, an agentic RAG framework that incorporates information sieving via LLM-as-a-knowledge-router. DeepSieve decomposes complex queries into structured sub-questions and recursively routes each to the most suitable knowledge source, filtering irrelevant information through a multi-stage distillation process. Our design emphasizes modularity, transparency, and adaptability, leveraging recent advances in agentic system design. Experiments on multi-hop QA tasks across heterogeneous sources demonstrate improved reasoning depth, retrieval precision, and interpretability over conventional RAG approaches. Our codes are available at https://github.com/MinghoKwok/DeepSieve.
RobotSmith: Generative Robotic Tool Design for Acquisition of Complex Manipulation Skills
Jun 17, 2025



Endowing robots with tool design abilities is critical for enabling them to solve complex manipulation tasks that would otherwise be intractable. While recent generative frameworks can automatically synthesize task settings, such as 3D scenes and reward functions, they have not yet addressed the challenge of tool-use scenarios. Simply retrieving human-designed tools might not be ideal since many tools (e.g., a rolling pin) are difficult for robotic manipulators to handle. Furthermore, existing tool design approaches either rely on predefined templates with limited parameter tuning or apply generic 3D generation methods that are not optimized for tool creation. To address these limitations, we propose RobotSmith, an automated pipeline that leverages the implicit physical knowledge embedded in vision-language models (VLMs) alongside the more accurate physics provided by physics simulations to design and use tools for robotic manipulation. Our system (1) iteratively proposes tool designs using collaborative VLM agents, (2) generates low-level robot trajectories for tool use, and (3) jointly optimizes tool geometry and usage for task performance. We evaluate our approach across a wide range of manipulation tasks involving rigid, deformable, and fluid objects. Experiments show that our method consistently outperforms strong baselines in terms of both task success rate and overall performance. Notably, our approach achieves a 50.0\% average success rate, significantly surpassing other baselines such as 3D generation (21.4%) and tool retrieval (11.1%). Finally, we deploy our system in real-world settings, demonstrating that the generated tools and their usage plans transfer effectively to physical execution, validating the practicality and generalization capabilities of our approach.
DeSocial: Blockchain-based Decentralized Social Networks
May 28, 2025Web 2.0 social platforms are inherently centralized, with user data and algorithmic decisions controlled by the platform. However, users can only passively receive social predictions without being able to choose the underlying algorithm, which limits personalization. Fortunately, with the emergence of blockchain, users are allowed to choose algorithms that are tailored to their local situation, improving prediction results in a personalized way. In a blockchain environment, each user possesses its own model to perform the social prediction, capturing different perspectives on social interactions. In our work, we propose DeSocial, a decentralized social network learning framework deployed on an Ethereum (ETH) local development chain that integrates distributed data storage, node-level consensus, and user-driven model selection through Ganache. In the first stage, each user leverages DeSocial to evaluate multiple backbone models on their local subgraph. DeSocial coordinates the execution and returns model-wise prediction results, enabling the user to select the most suitable backbone for personalized social prediction. Then, DeSocial uniformly selects several validation nodes that possess the algorithm specified by each user, and aggregates the prediction results by majority voting, to prevent errors caused by any single model's misjudgment. Extensive experiments show that DeSocial has an evident improvement compared to the five classical centralized social network learning models, promoting user empowerment in blockchain-based decentralized social networks, showing the importance of multi-node validation and personalized algorithm selection based on blockchain. Our implementation is available at: https://github.com/agiresearch/DeSocial.
FlashBias: Fast Computation of Attention with Bias
May 17, 2025Attention mechanism has emerged as a foundation module of modern deep learning models and has also empowered many milestones in various domains. Moreover, FlashAttention with IO-aware speedup resolves the efficiency issue of standard attention, further promoting its practicality. Beyond canonical attention, attention with bias also widely exists, such as relative position bias in vision and language models and pair representation bias in AlphaFold. In these works, prior knowledge is introduced as an additive bias term of attention weights to guide the learning process, which has been proven essential for model performance. Surprisingly, despite the common usage of attention with bias, its targeted efficiency optimization is still absent, which seriously hinders its wide applications in complex tasks. Diving into the computation of FlashAttention, we prove that its optimal efficiency is determined by the rank of the attention weight matrix. Inspired by this theoretical result, this paper presents FlashBias based on the low-rank compressed sensing theory, which can provide fast-exact computation for many widely used attention biases and a fast-accurate approximation for biases in general formalization. FlashBias can fully take advantage of the extremely optimized matrix multiplication operation in modern GPUs, achieving 1.5$\times$ speedup for AlphaFold, and over 2$\times$ speedup for attention with bias in vision and language models without loss of accuracy.
A0: An Affordance-Aware Hierarchical Model for General Robotic Manipulation
Apr 21, 2025



Robotic manipulation faces critical challenges in understanding spatial affordances--the "where" and "how" of object interactions--essential for complex manipulation tasks like wiping a board or stacking objects. Existing methods, including modular-based and end-to-end approaches, often lack robust spatial reasoning capabilities. Unlike recent point-based and flow-based affordance methods that focus on dense spatial representations or trajectory modeling, we propose A0, a hierarchical affordance-aware diffusion model that decomposes manipulation tasks into high-level spatial affordance understanding and low-level action execution. A0 leverages the Embodiment-Agnostic Affordance Representation, which captures object-centric spatial affordances by predicting contact points and post-contact trajectories. A0 is pre-trained on 1 million contact points data and fine-tuned on annotated trajectories, enabling generalization across platforms. Key components include Position Offset Attention for motion-aware feature extraction and a Spatial Information Aggregation Layer for precise coordinate mapping. The model's output is executed by the action execution module. Experiments on multiple robotic systems (Franka, Kinova, Realman, and Dobot) demonstrate A0's superior performance in complex tasks, showcasing its efficiency, flexibility, and real-world applicability.
LLM as GNN: Graph Vocabulary Learning for Text-Attributed Graph Foundation Models
Mar 05, 2025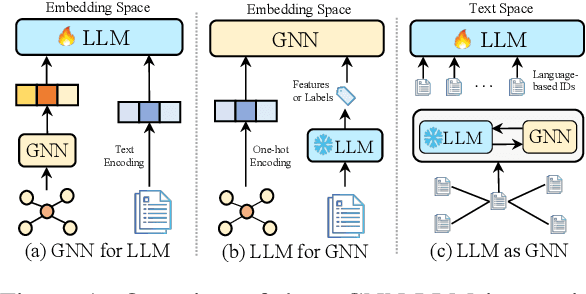
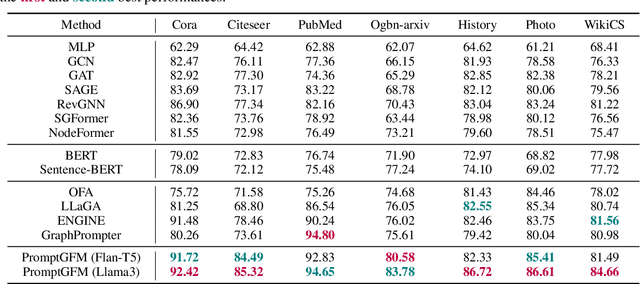
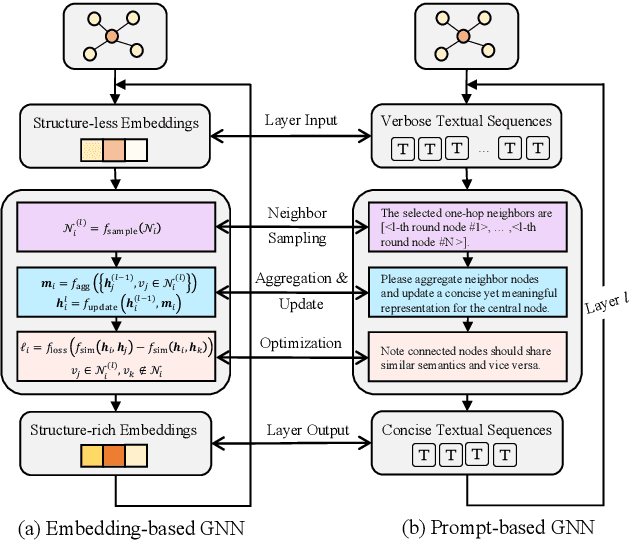
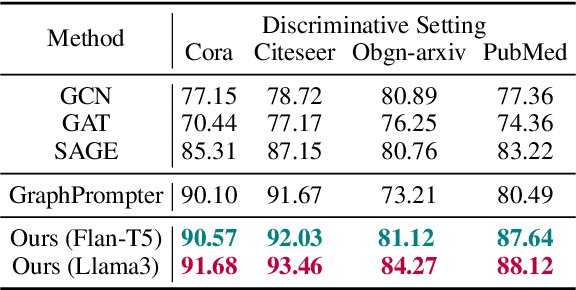
Text-Attributed Graphs (TAGs), where each node is associated with text descriptions, are ubiquitous in real-world scenarios. They typically exhibit distinctive structure and domain-specific knowledge, motivating the development of a Graph Foundation Model (GFM) that generalizes across diverse graphs and tasks. Despite large efforts to integrate Large Language Models (LLMs) and Graph Neural Networks (GNNs) for TAGs, existing approaches suffer from decoupled architectures with two-stage alignment, limiting their synergistic potential. Even worse, existing methods assign out-of-vocabulary (OOV) tokens to graph nodes, leading to graph-specific semantics, token explosion, and incompatibility with task-oriented prompt templates, which hinders cross-graph and cross-task transferability. To address these challenges, we propose PromptGFM, a versatile GFM for TAGs grounded in graph vocabulary learning. PromptGFM comprises two key components: (1) Graph Understanding Module, which explicitly prompts LLMs to replicate the finest GNN workflow within the text space, facilitating seamless GNN-LLM integration and elegant graph-text alignment; (2) Graph Inference Module, which establishes a language-based graph vocabulary ensuring expressiveness, transferability, and scalability, enabling readable instructions for LLM fine-tuning. Extensive experiments demonstrate our superiority and transferability across diverse graphs and tasks. The code is available at this: https://github.com/agiresearch/PromptGFM.
Two-Stage Pretraining for Molecular Property Prediction in the Wild
Nov 05, 2024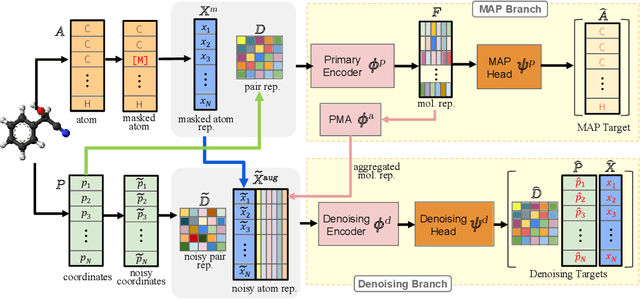

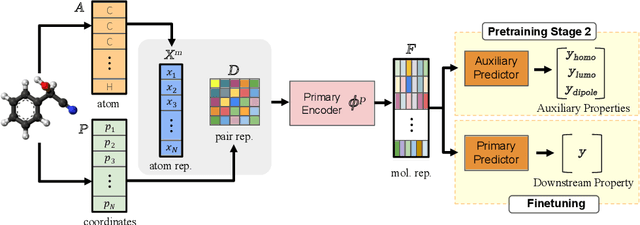
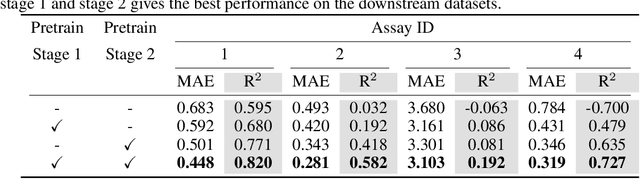
Accurate property prediction is crucial for accelerating the discovery of new molecules. Although deep learning models have achieved remarkable success, their performance often relies on large amounts of labeled data that are expensive and time-consuming to obtain. Thus, there is a growing need for models that can perform well with limited experimentally-validated data. In this work, we introduce MoleVers, a versatile pretrained model designed for various types of molecular property prediction in the wild, i.e., where experimentally-validated molecular property labels are scarce. MoleVers adopts a two-stage pretraining strategy. In the first stage, the model learns molecular representations from large unlabeled datasets via masked atom prediction and dynamic denoising, a novel task enabled by a new branching encoder architecture. In the second stage, MoleVers is further pretrained using auxiliary labels obtained with inexpensive computational methods, enabling supervised learning without the need for costly experimental data. This two-stage framework allows MoleVers to learn representations that generalize effectively across various downstream datasets. We evaluate MoleVers on a new benchmark comprising 22 molecular datasets with diverse types of properties, the majority of which contain 50 or fewer training labels reflecting real-world conditions. MoleVers achieves state-of-the-art results on 20 out of the 22 datasets, and ranks second among the remaining two, highlighting its ability to bridge the gap between data-hungry models and real-world conditions where practically-useful labels are scarce.