 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDo Sparse Autoencoders Generalize? A Case Study of Answerability
Feb 27, 2025Sparse autoencoders (SAEs) have emerged as a promising approach in language model interpretability, offering unsupervised extraction of sparse features. For interpretability methods to succeed, they must identify abstract features across domains, and these features can often manifest differently in each context. We examine this through "answerability"-a model's ability to recognize answerable questions. We extensively evaluate SAE feature generalization across diverse answerability datasets for Gemma 2 SAEs. Our analysis reveals that residual stream probes outperform SAE features within domains, but generalization performance differs sharply. SAE features demonstrate inconsistent transfer ability, and residual stream probes similarly show high variance out of distribution. Overall, this demonstrates the need for quantitative methods to predict feature generalization in SAE-based interpretability.
Representing Molecules as Random Walks Over Interpretable Grammars
Mar 13, 2024



Recent research in molecular discovery has primarily been devoted to small, drug-like molecules, leaving many similarly important applications in material design without adequate technology. These applications often rely on more complex molecular structures with fewer examples that are carefully designed using known substructures. We propose a data-efficient and interpretable model for representing and reasoning over such molecules in terms of graph grammars that explicitly describe the hierarchical design space featuring motifs to be the design basis. We present a novel representation in the form of random walks over the design space, which facilitates both molecule generation and property prediction. We demonstrate clear advantages over existing methods in terms of performance, efficiency, and synthesizability of predicted molecules, and we provide detailed insights into the method's chemical interpretability.
Improving Self-supervised Molecular Representation Learning using Persistent Homology
Nov 29, 2023Self-supervised learning (SSL) has great potential for molecular representation learning given the complexity of molecular graphs, the large amounts of unlabelled data available, the considerable cost of obtaining labels experimentally, and the hence often only small training datasets. The importance of the topic is reflected in the variety of paradigms and architectures that have been investigated recently. Yet the differences in performance seem often minor and are barely understood to date. In this paper, we study SSL based on persistent homology (PH), a mathematical tool for modeling topological features of data that persist across multiple scales. It has several unique features which particularly suit SSL, naturally offering: different views of the data, stability in terms of distance preservation, and the opportunity to flexibly incorporate domain knowledge. We (1) investigate an autoencoder, which shows the general representational power of PH, and (2) propose a contrastive loss that complements existing approaches. We rigorously evaluate our approach for molecular property prediction and demonstrate its particular features in improving the embedding space: after SSL, the representations are better and offer considerably more predictive power than the baselines over different probing tasks; our loss increases baseline performance, sometimes largely; and we often obtain substantial improvements over very small datasets, a common scenario in practice.
Hierarchical Grammar-Induced Geometry for Data-Efficient Molecular Property Prediction
Sep 04, 2023The prediction of molecular properties is a crucial task in the field of material and drug discovery. The potential benefits of using deep learning techniques are reflected in the wealth of recent literature. Still, these techniques are faced with a common challenge in practice: Labeled data are limited by the cost of manual extraction from literature and laborious experimentation. In this work, we propose a data-efficient property predictor by utilizing a learnable hierarchical molecular grammar that can generate molecules from grammar production rules. Such a grammar induces an explicit geometry of the space of molecular graphs, which provides an informative prior on molecular structural similarity. The property prediction is performed using graph neural diffusion over the grammar-induced geometry. On both small and large datasets, our evaluation shows that this approach outperforms a wide spectrum of baselines, including supervised and pre-trained graph neural networks. We include a detailed ablation study and further analysis of our solution, showing its effectiveness in cases with extremely limited data. Code is available at https://github.com/gmh14/Geo-DEG.
Data-Efficient Graph Grammar Learning for Molecular Generation
Mar 15, 2022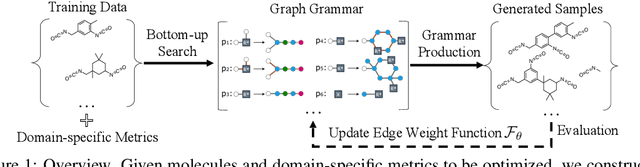
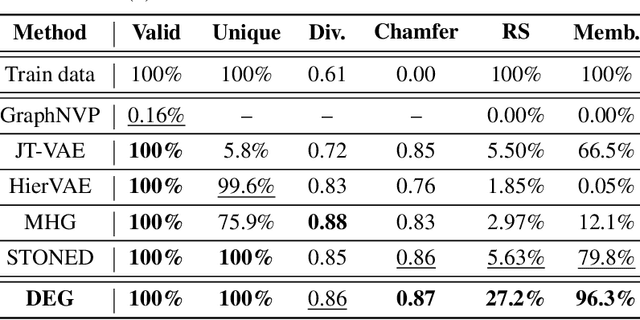
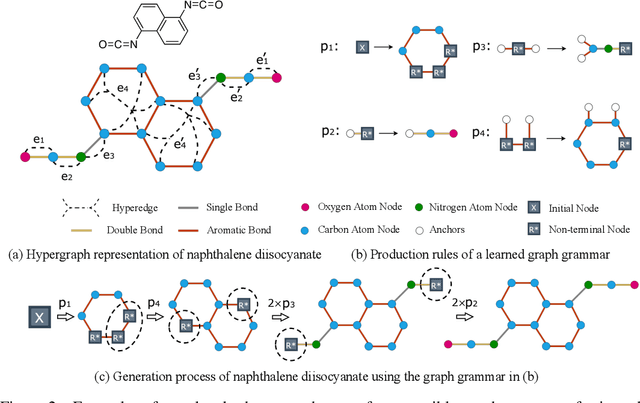
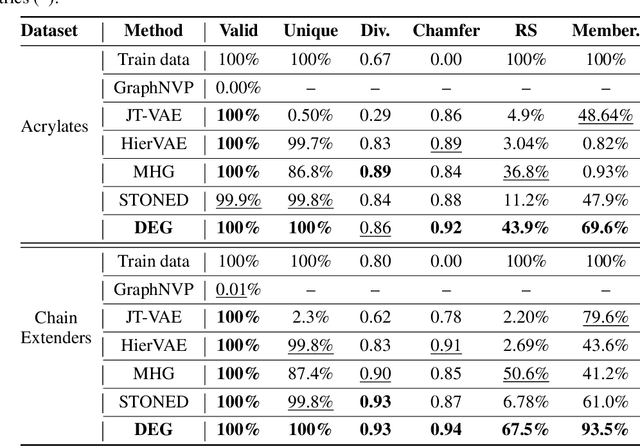
The problem of molecular generation has received significant attention recently. Existing methods are typically based on deep neural networks and require training on large datasets with tens of thousands of samples. In practice, however, the size of class-specific chemical datasets is usually limited (e.g., dozens of samples) due to labor-intensive experimentation and data collection. This presents a considerable challenge for the deep learning generative models to comprehensively describe the molecular design space. Another major challenge is to generate only physically synthesizable molecules. This is a non-trivial task for neural network-based generative models since the relevant chemical knowledge can only be extracted and generalized from the limited training data. In this work, we propose a data-efficient generative model that can be learned from datasets with orders of magnitude smaller sizes than common benchmarks. At the heart of this method is a learnable graph grammar that generates molecules from a sequence of production rules. Without any human assistance, these production rules are automatically constructed from training data. Furthermore, additional chemical knowledge can be incorporated in the model by further grammar optimization. Our learned graph grammar yields state-of-the-art results on generating high-quality molecules for three monomer datasets that contain only ${\sim}20$ samples each. Our approach also achieves remarkable performance in a challenging polymer generation task with only $117$ training samples and is competitive against existing methods using $81$k data points. Code is available at https://github.com/gmh14/data_efficient_grammar.
Software Vulnerability Detection via Deep Learning over Disaggregated Code Graph Representation
Sep 07, 2021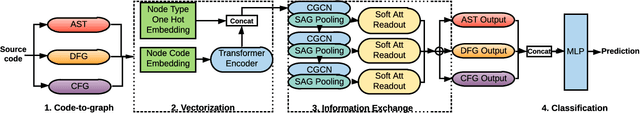
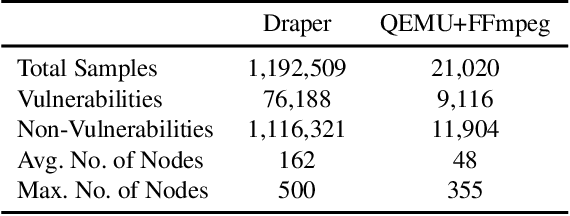
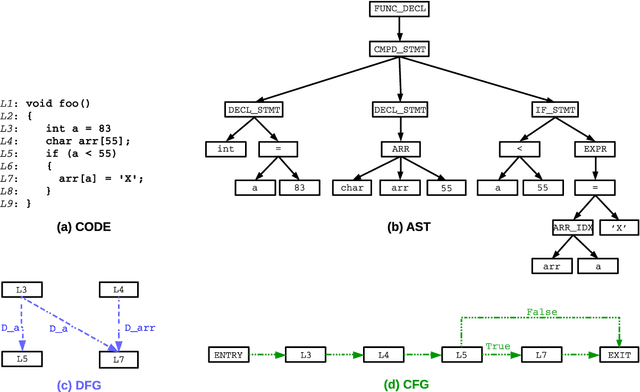
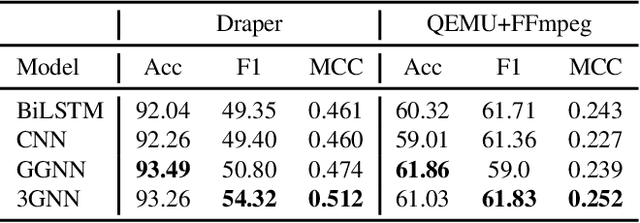
Identifying vulnerable code is a precautionary measure to counter software security breaches. Tedious expert effort has been spent to build static analyzers, yet insecure patterns are barely fully enumerated. This work explores a deep learning approach to automatically learn the insecure patterns from code corpora. Because code naturally admits graph structures with parsing, we develop a novel graph neural network (GNN) to exploit both the semantic context and structural regularity of a program, in order to improve prediction performance. Compared with a generic GNN, our enhancements include a synthesis of multiple representations learned from the several parsed graphs of a program, and a new training loss metric that leverages the fine granularity of labeling. Our model outperforms multiple text, image and graph-based approaches, across two real-world datasets.
Improving Inductive Link Prediction Using Hyper-Relational Facts
Jul 10, 2021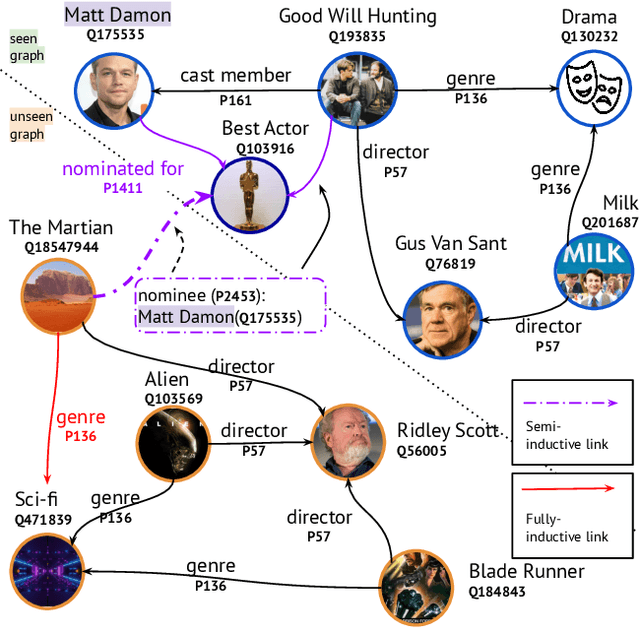

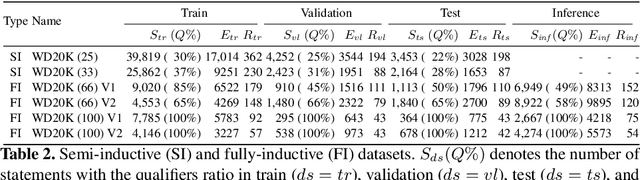
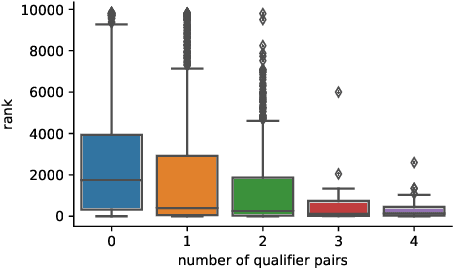
For many years, link prediction on knowledge graphs (KGs) has been a purely transductive task, not allowing for reasoning on unseen entities. Recently, increasing efforts are put into exploring semi- and fully inductive scenarios, enabling inference over unseen and emerging entities. Still, all these approaches only consider triple-based \glspl{kg}, whereas their richer counterparts, hyper-relational KGs (e.g., Wikidata), have not yet been properly studied. In this work, we classify different inductive settings and study the benefits of employing hyper-relational KGs on a wide range of semi- and fully inductive link prediction tasks powered by recent advancements in graph neural networks. Our experiments on a novel set of benchmarks show that qualifiers over typed edges can lead to performance improvements of 6% of absolute gains (for the Hits@10 metric) compared to triple-only baselines. Our code is available at \url{https://github.com/mali-git/hyper_relational_ilp}.
Relation Matters in Sampling: A Scalable Multi-Relational Graph Neural Network for Drug-Drug Interaction Prediction
May 28, 2021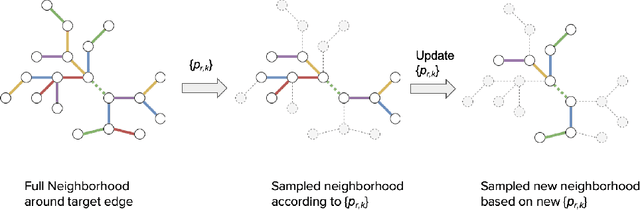


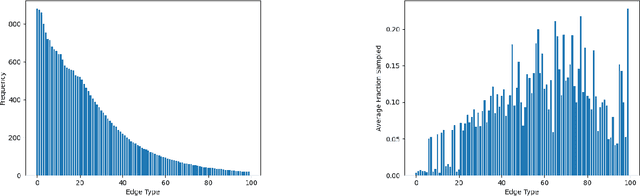
Sampling is an established technique to scale graph neural networks to large graphs. Current approaches however assume the graphs to be homogeneous in terms of relations and ignore relation types, critically important in biomedical graphs. Multi-relational graphs contain various types of relations that usually come with variable frequency and have different importance for the problem at hand. We propose an approach to modeling the importance of relation types for neighborhood sampling in graph neural networks and show that we can learn the right balance: relation-type probabilities that reflect both frequency and importance. Our experiments on drug-drug interaction prediction show that state-of-the-art graph neural networks profit from relation-dependent sampling in terms of both accuracy and efficiency.
Project CodeNet: A Large-Scale AI for Code Dataset for Learning a Diversity of Coding Tasks
May 25, 2021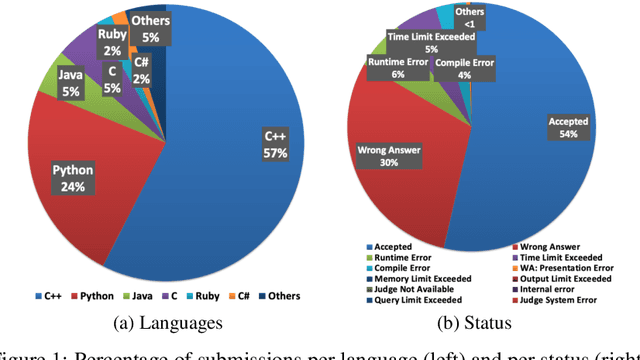
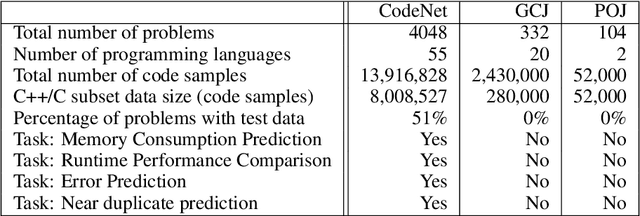
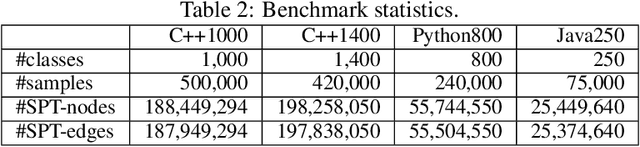
Advancements in deep learning and machine learning algorithms have enabled breakthrough progress in computer vision, speech recognition, natural language processing and beyond. In addition, over the last several decades, software has been built into the fabric of every aspect of our society. Together, these two trends have generated new interest in the fast-emerging research area of AI for Code. As software development becomes ubiquitous across all industries and code infrastructure of enterprise legacy applications ages, it is more critical than ever to increase software development productivity and modernize legacy applications. Over the last decade, datasets like ImageNet, with its large scale and diversity, have played a pivotal role in algorithmic advancements from computer vision to language and speech understanding. In this paper, we present Project CodeNet, a first-of-its-kind, very large scale, diverse, and high-quality dataset to accelerate the algorithmic advancements in AI for Code. It consists of 14M code samples and about 500M lines of code in 55 different programming languages. Project CodeNet is not only unique in its scale, but also in the diversity of coding tasks it can help benchmark: from code similarity and classification for advances in code recommendation algorithms, and code translation between a large variety programming languages, to advances in code performance (both runtime, and memory) improvement techniques. CodeNet also provides sample input and output test sets for over 7M code samples, which can be critical for determining code equivalence in different languages. As a usability feature, we provide several preprocessing tools in Project CodeNet to transform source codes into representations that can be readily used as inputs into machine learning models.
Directed Acyclic Graph Neural Networks
Feb 02, 2021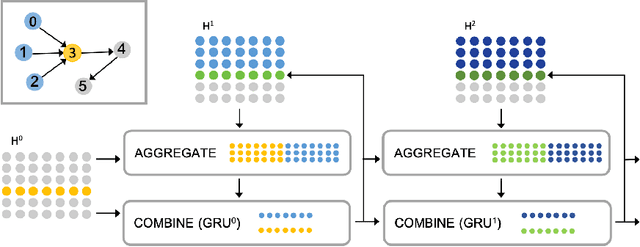
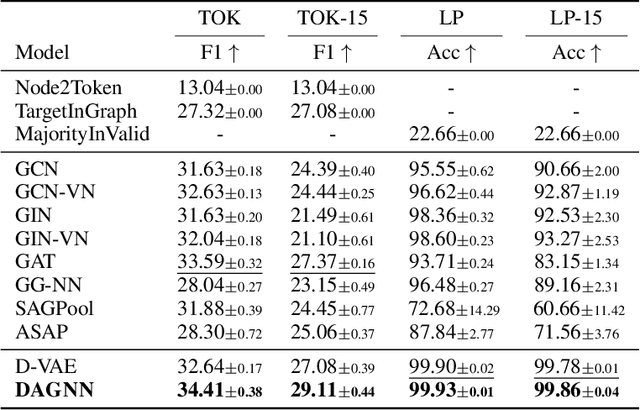
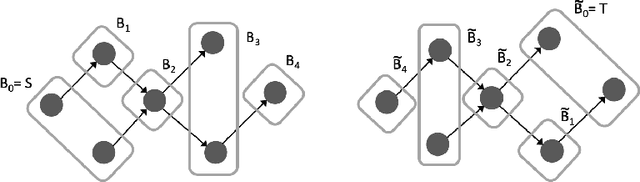

Graph-structured data ubiquitously appears in science and engineering. Graph neural networks (GNNs) are designed to exploit the relational inductive bias exhibited in graphs; they have been shown to outperform other forms of neural networks in scenarios where structure information supplements node features. The most common GNN architecture aggregates information from neighborhoods based on message passing. Its generality has made it broadly applicable. In this paper, we focus on a special, yet widely used, type of graphs -- DAGs -- and inject a stronger inductive bias -- partial ordering -- into the neural network design. We propose the \emph{directed acyclic graph neural network}, DAGNN, an architecture that processes information according to the flow defined by the partial order. DAGNN can be considered a framework that entails earlier works as special cases (e.g., models for trees and models updating node representations recurrently), but we identify several crucial components that prior architectures lack. We perform comprehensive experiments, including ablation studies, on representative DAG datasets (i.e., source code, neural architectures, and probabilistic graphical models) and demonstrate the superiority of DAGNN over simpler DAG architectures as well as general graph architectures.