 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFinGAIA: An End-to-End Benchmark for Evaluating AI Agents in Finance
Jul 23, 2025



The booming development of AI agents presents unprecedented opportunities for automating complex tasks across various domains. However, their multi-step, multi-tool collaboration capabilities in the financial sector remain underexplored. This paper introduces FinGAIA, an end-to-end benchmark designed to evaluate the practical abilities of AI agents in the financial domain. FinGAIA comprises 407 meticulously crafted tasks, spanning seven major financial sub-domains: securities, funds, banking, insurance, futures, trusts, and asset management. These tasks are organized into three hierarchical levels of scenario depth: basic business analysis, asset decision support, and strategic risk management. We evaluated 10 mainstream AI agents in a zero-shot setting. The best-performing agent, ChatGPT, achieved an overall accuracy of 48.9\%, which, while superior to non-professionals, still lags financial experts by over 35 percentage points. Error analysis has revealed five recurring failure patterns: Cross-modal Alignment Deficiency, Financial Terminological Bias, Operational Process Awareness Barrier, among others. These patterns point to crucial directions for future research. Our work provides the first agent benchmark closely related to the financial domain, aiming to objectively assess and promote the development of agents in this crucial field. Partial data is available at https://github.com/SUFE-AIFLM-Lab/FinGAIA.
Enhanced Multimodal Aspect-Based Sentiment Analysis by LLM-Generated Rationales
May 20, 2025There has been growing interest in Multimodal Aspect-Based Sentiment Analysis (MABSA) in recent years. Existing methods predominantly rely on pre-trained small language models (SLMs) to collect information related to aspects and sentiments from both image and text, with an aim to align these two modalities. However, small SLMs possess limited capacity and knowledge, often resulting in inaccurate identification of meaning, aspects, sentiments, and their interconnections in textual and visual data. On the other hand, Large language models (LLMs) have shown exceptional capabilities in various tasks by effectively exploring fine-grained information in multimodal data. However, some studies indicate that LLMs still fall short compared to fine-tuned small models in the field of ABSA. Based on these findings, we propose a novel framework, termed LRSA, which combines the decision-making capabilities of SLMs with additional information provided by LLMs for MABSA. Specifically, we inject explanations generated by LLMs as rationales into SLMs and employ a dual cross-attention mechanism for enhancing feature interaction and fusion, thereby augmenting the SLMs' ability to identify aspects and sentiments. We evaluated our method using two baseline models, numerous experiments highlight the superiority of our approach on three widely-used benchmarks, indicating its generalizability and applicability to most pre-trained models for MABSA.
DeepGDel: Deep Learning-based Gene Deletion Prediction Framework for Growth-Coupled Production in Genome-Scale Metabolic Models
Apr 08, 2025

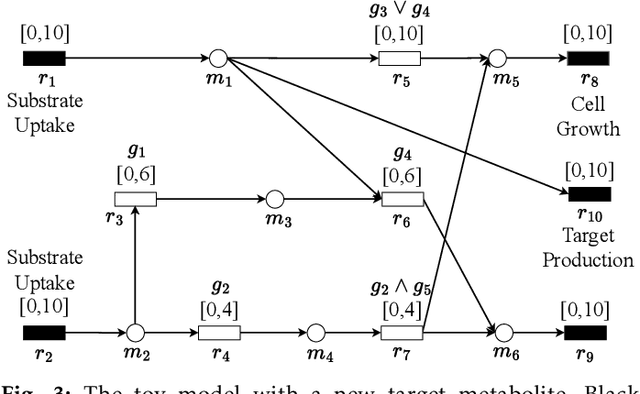

In genome-scale constraint-based metabolic models, gene deletion strategies are crucial for achieving growth-coupled production, where cell growth and target metabolite production are simultaneously achieved. While computational methods for calculating gene deletions have been widely explored and contribute to developing gene deletion strategy databases, current approaches are limited in leveraging new data-driven paradigms, such as machine learning, for more efficient strain design. Therefore, it is necessary to propose a fundamental framework for this objective. In this study, we first formulate the problem of gene deletion strategy prediction and then propose a framework for predicting gene deletion strategies for growth-coupled production in genome-scale metabolic models. The proposed framework leverages deep learning algorithms to learn and integrate sequential gene and metabolite data representation, enabling the automatic gene deletion strategy prediction. Computational experiment results demonstrate the feasibility of the proposed framework, showing substantial improvements over the baseline method. Specifically, the proposed framework achieves a 17.64%, 27.15%, and 18.07% increase in overall accuracy across three metabolic models of different scales under study, while maintaining balanced precision and recall in predicting gene deletion statuses. The source code and examples for the framework are publicly available at https://github.com/MetNetComp/DeepGDel.
Fin-R1: A Large Language Model for Financial Reasoning through Reinforcement Learning
Mar 21, 2025



Reasoning large language models are rapidly evolving across various domains. However, their capabilities in handling complex financial tasks still require in-depth exploration. In this paper, we introduce Fin-R1, a reasoning large language model specifically designed for the financial sector. Fin-R1 is built using a two-stage architecture, leveraging a financial reasoning dataset distilled and processed based on DeepSeek-R1. Through supervised fine-tuning (SFT) and reinforcement learning (RL) training, it demonstrates performance close to DeepSeek-R1 with a parameter size of 7 billion across a range of financial reasoning tasks. It achieves the state-of-the-art (SOTA) in the FinQA and ConvFinQA tasks between those LLMs in our evaluation, surpassing larger models in other tasks as well. Fin-R1 showcases strong reasoning and decision-making capabilities, providing solutions to various problems encountered in the financial domain. Our code is available at https://github.com/SUFE-AIFLM-Lab/Fin-R1.
ExPath: Towards Explaining Targeted Pathways for Biological Knowledge Bases
Feb 25, 2025Biological knowledge bases provide systemically functional pathways of cells or organisms in terms of molecular interaction. However, recognizing more targeted pathways, particularly when incorporating wet-lab experimental data, remains challenging and typically requires downstream biological analyses and expertise. In this paper, we frame this challenge as a solvable graph learning and explaining task and propose a novel pathway inference framework, ExPath, that explicitly integrates experimental data, specifically amino acid sequences (AA-seqs), to classify various graphs (bio-networks) in biological databases. The links (representing pathways) that contribute more to classification can be considered as targeted pathways. Technically, ExPath comprises three components: (1) a large protein language model (pLM) that encodes and embeds AA-seqs into graph, overcoming traditional obstacles in processing AA-seq data, such as BLAST; (2) PathMamba, a hybrid architecture combining graph neural networks (GNNs) with state-space sequence modeling (Mamba) to capture both local interactions and global pathway-level dependencies; and (3) PathExplainer, a subgraph learning module that identifies functionally critical nodes and edges through trainable pathway masks. We also propose ML-oriented biological evaluations and a new metric. The experiments involving 301 bio-networks evaluations demonstrate that pathways inferred by ExPath maintain biological meaningfulness. We will publicly release curated 301 bio-network data soon.
Can Large Language Models Replace Data Scientists in Clinical Research?
Oct 28, 2024



Data science plays a critical role in clinical research, but it requires professionals with expertise in coding and medical data analysis. Large language models (LLMs) have shown great potential in supporting medical tasks and performing well in general coding tests. However, these tests do not assess LLMs' ability to handle data science tasks in medicine, nor do they explore their practical utility in clinical research. To address this, we developed a dataset consisting of 293 real-world data science coding tasks, based on 39 published clinical studies, covering 128 tasks in Python and 165 tasks in R. This dataset simulates realistic clinical research scenarios using patient data. Our findings reveal that cutting-edge LLMs struggle to generate perfect solutions, frequently failing to follow input instructions, understand target data, and adhere to standard analysis practices. Consequently, LLMs are not yet ready to fully automate data science tasks. We benchmarked advanced adaptation methods and found two to be particularly effective: chain-of-thought prompting, which provides a step-by-step plan for data analysis, which led to a 60% improvement in code accuracy; and self-reflection, enabling LLMs to iteratively refine their code, yielding a 38% accuracy improvement. Building on these insights, we developed a platform that integrates LLMs into the data science workflow for medical professionals. In a user study with five medical doctors, we found that while LLMs cannot fully automate coding tasks, they significantly streamline the programming process. We found that 80% of their submitted code solutions were incorporated from LLM-generated code, with up to 96% reuse in some cases. Our analysis highlights the potential of LLMs, when integrated into expert workflows, to enhance data science efficiency in clinical research.
GeSubNet: Gene Interaction Inference for Disease Subtype Network Generation
Oct 17, 2024


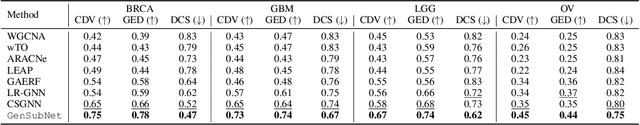
Retrieving gene functional networks from knowledge databases presents a challenge due to the mismatch between disease networks and subtype-specific variations. Current solutions, including statistical and deep learning methods, often fail to effectively integrate gene interaction knowledge from databases or explicitly learn subtype-specific interactions. To address this mismatch, we propose GeSubNet, which learns a unified representation capable of predicting gene interactions while distinguishing between different disease subtypes. Graphs generated by such representations can be considered subtype-specific networks. GeSubNet is a multi-step representation learning framework with three modules: First, a deep generative model learns distinct disease subtypes from patient gene expression profiles. Second, a graph neural network captures representations of prior gene networks from knowledge databases, ensuring accurate physical gene interactions. Finally, we integrate these two representations using an inference loss that leverages graph generation capabilities, conditioned on the patient separation loss, to refine subtype-specific information in the learned representation. GeSubNet consistently outperforms traditional methods, with average improvements of 30.6%, 21.0%, 20.1%, and 56.6% across four graph evaluation metrics, averaged over four cancer datasets. Particularly, we conduct a biological simulation experiment to assess how the behavior of selected genes from over 11,000 candidates affects subtypes or patient distributions. The results show that the generated network has the potential to identify subtype-specific genes with an 83% likelihood of impacting patient distribution shifts. The GeSubNet resource is available: https://anonymous.4open.science/r/GeSubNet/
CMOB: Large-Scale Cancer Multi-Omics Benchmark with Open Datasets, Tasks, and Baselines
Sep 02, 2024Machine learning has shown great potential in the field of cancer multi-omics studies, offering incredible opportunities for advancing precision medicine. However, the challenges associated with dataset curation and task formulation pose significant hurdles, especially for researchers lacking a biomedical background. Here, we introduce the CMOB, the first large-scale cancer multi-omics benchmark integrates the TCGA platform, making data resources accessible and usable for machine learning researchers without significant preparation and expertise.To date, CMOB includes a collection of 20 cancer multi-omics datasets covering 32 cancers, accompanied by a systematic data processing pipeline. CMOB provides well-processed dataset versions to support 20 meaningful tasks in four studies, with a collection of benchmarks. We also integrate CMOB with two complementary resources and various biological tools to explore broader research avenues.All resources are open-accessible with user-friendly and compatible integration scripts that enable non-experts to easily incorporate this complementary information for various tasks. We conduct extensive experiments on selected datasets to offer recommendations on suitable machine learning baselines for specific applications. Through CMOB, we aim to facilitate algorithmic advances and hasten the development, validation, and clinical translation of machine-learning models for personalized cancer treatments. CMOB is available on GitHub (\url{https://github.com/chenzRG/Cancer-Multi-Omics-Benchmark}).
A Bounding Box is Worth One Token: Interleaving Layout and Text in a Large Language Model for Document Understanding
Jul 02, 2024



Recently, many studies have demonstrated that exclusively incorporating OCR-derived text and spatial layouts with large language models (LLMs) can be highly effective for document understanding tasks. However, existing methods that integrate spatial layouts with text have limitations, such as producing overly long text sequences or failing to fully leverage the autoregressive traits of LLMs. In this work, we introduce Interleaving Layout and Text in a Large Language Model (LayTextLLM)} for document understanding. In particular, LayTextLLM projects each bounding box to a single embedding and interleaves it with text, efficiently avoiding long sequence issues while leveraging autoregressive traits of LLMs. LayTextLLM not only streamlines the interaction of layout and textual data but also shows enhanced performance in Key Information Extraction (KIE) and Visual Question Answering (VQA). Comprehensive benchmark evaluations reveal significant improvements, with a 27.0% increase on KIE tasks and 24.1% on VQA tasks compared to previous state-of-the-art document understanding MLLMs, as well as a 15.5% improvement over other SOTA OCR-based LLMs on KIE tasks.
PaDeLLM-NER: Parallel Decoding in Large Language Models for Named Entity Recognition
Feb 15, 2024
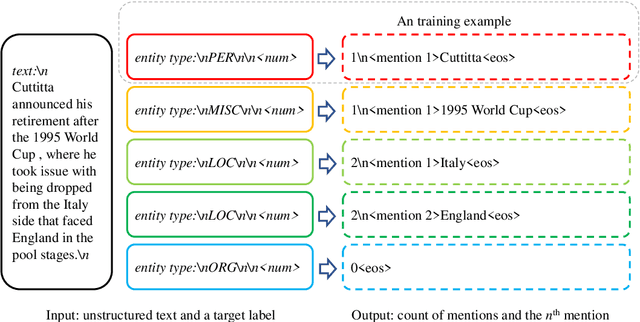
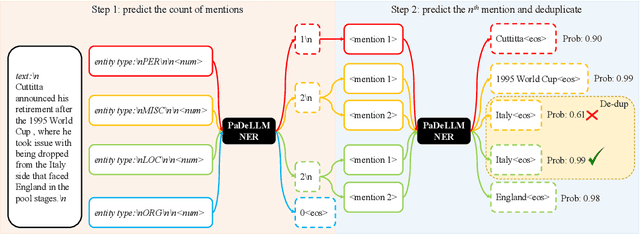
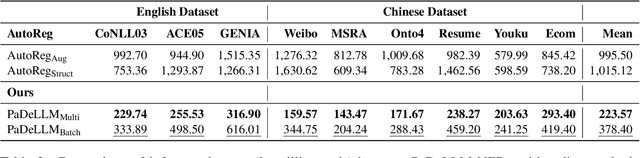
In this study, we aim to reduce generation latency for Named Entity Recognition (NER) with Large Language Models (LLMs). The main cause of high latency in LLMs is the sequential decoding process, which autoregressively generates all labels and mentions for NER, significantly increase the sequence length. To this end, we introduce Parallel Decoding in LLM for NE} (PaDeLLM-NER), a approach that integrates seamlessly into existing generative model frameworks without necessitating additional modules or architectural modifications. PaDeLLM-NER allows for the simultaneous decoding of all mentions, thereby reducing generation latency. Experiments reveal that PaDeLLM-NER significantly increases inference speed that is 1.76 to 10.22 times faster than the autoregressive approach for both English and Chinese. Simultaneously it maintains the quality of predictions as evidenced by the performance that is on par with the state-of-the-art across various datasets.