 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOptimizing EEG Graph Structure for Seizure Detection: An Information Bottleneck and Self-Supervised Learning Approach
Apr 02, 2026Seizure detection from EEG signals is highly challenging due to complex spatiotemporal dynamics and extreme inter-patient variability. To model them, recent methods construct dynamic graphs via statistical correlations, predefined similarity measures, or implicit learning, yet rarely account for EEG's noisy nature. Consequently, these graphs usually contain redundant or task-irrelevant connections, undermining model performance even with state-of-the-art architectures. In this paper, we present a new perspective for EEG seizure detection: jointly learning denoised dynamic graph structures and informative spatial-temporal representations guided by the Information Bottleneck (IB). Unlike prior approaches, our graph constructor explicitly accounts for the noisy characteristics of EEG data, producing compact and reliable connectivity patterns that better support downstream seizure detection. To further enhance representation learning, we employ a self-supervised Graph Masked AutoEncoder that reconstructs masked EEG signals based on dynamic graph context, promoting structure-aware and compact representations aligned with the IB principle. Bringing things together, we introduce Information Bottleneck-guided EEG SeizuRE DetectioN via SElf-Supervised Learning (IRENE), which explicitly learns dynamic graph structures and interpretable spatial-temporal EEG representations. IRENE addresses three core challenges: (i) Identifying the most informative nodes and edges; (ii) Explaining seizure propagation in the brain network; and (iii) Enhancing robustness against label scarcity and inter-patient variability. Extensive experiments on benchmark EEG datasets demonstrate that our method outperforms state-of-the-art baselines in seizure detection and provides clinically meaningful insights into seizure dynamics. The source code is available at https://github.com/LabRAI/IRENE.
ODEBrain: Continuous-Time EEG Graph for Modeling Dynamic Brain Networks
Feb 26, 2026Modeling neural population dynamics is crucial for foundational neuroscientific research and various clinical applications. Conventional latent variable methods typically model continuous brain dynamics through discretizing time with recurrent architecture, which necessarily results in compounded cumulative prediction errors and failure of capturing instantaneous, nonlinear characteristics of EEGs. We propose ODEBRAIN, a Neural ODE latent dynamic forecasting framework to overcome these challenges by integrating spatio-temporal-frequency features into spectral graph nodes, followed by a Neural ODE modeling the continuous latent dynamics. Our design ensures that latent representations can capture stochastic variations of complex brain states at any given time point. Extensive experiments verify that ODEBRAIN can improve significantly over existing methods in forecasting EEG dynamics with enhanced robustness and generalization capabilities.
EvoBrain: Dynamic Multi-channel EEG Graph Modeling for Time-evolving Brain Network
Sep 19, 2025Dynamic GNNs, which integrate temporal and spatial features in Electroencephalography (EEG) data, have shown great potential in automating seizure detection. However, fully capturing the underlying dynamics necessary to represent brain states, such as seizure and non-seizure, remains a non-trivial task and presents two fundamental challenges. First, most existing dynamic GNN methods are built on temporally fixed static graphs, which fail to reflect the evolving nature of brain connectivity during seizure progression. Second, current efforts to jointly model temporal signals and graph structures and, more importantly, their interactions remain nascent, often resulting in inconsistent performance. To address these challenges, we present the first theoretical analysis of these two problems, demonstrating the effectiveness and necessity of explicit dynamic modeling and time-then-graph dynamic GNN method. Building on these insights, we propose EvoBrain, a novel seizure detection model that integrates a two-stream Mamba architecture with a GCN enhanced by Laplacian Positional Encoding, following neurological insights. Moreover, EvoBrain incorporates explicitly dynamic graph structures, allowing both nodes and edges to evolve over time. Our contributions include (a) a theoretical analysis proving the expressivity advantage of explicit dynamic modeling and time-then-graph over other approaches, (b) a novel and efficient model that significantly improves AUROC by 23% and F1 score by 30%, compared with the dynamic GNN baseline, and (c) broad evaluations of our method on the challenging early seizure prediction tasks.
ExPath: Towards Explaining Targeted Pathways for Biological Knowledge Bases
Feb 25, 2025Biological knowledge bases provide systemically functional pathways of cells or organisms in terms of molecular interaction. However, recognizing more targeted pathways, particularly when incorporating wet-lab experimental data, remains challenging and typically requires downstream biological analyses and expertise. In this paper, we frame this challenge as a solvable graph learning and explaining task and propose a novel pathway inference framework, ExPath, that explicitly integrates experimental data, specifically amino acid sequences (AA-seqs), to classify various graphs (bio-networks) in biological databases. The links (representing pathways) that contribute more to classification can be considered as targeted pathways. Technically, ExPath comprises three components: (1) a large protein language model (pLM) that encodes and embeds AA-seqs into graph, overcoming traditional obstacles in processing AA-seq data, such as BLAST; (2) PathMamba, a hybrid architecture combining graph neural networks (GNNs) with state-space sequence modeling (Mamba) to capture both local interactions and global pathway-level dependencies; and (3) PathExplainer, a subgraph learning module that identifies functionally critical nodes and edges through trainable pathway masks. We also propose ML-oriented biological evaluations and a new metric. The experiments involving 301 bio-networks evaluations demonstrate that pathways inferred by ExPath maintain biological meaningfulness. We will publicly release curated 301 bio-network data soon.
GeSubNet: Gene Interaction Inference for Disease Subtype Network Generation
Oct 17, 2024


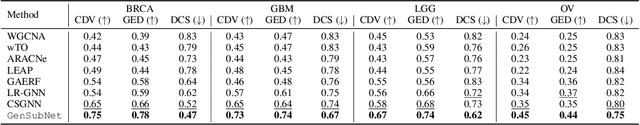
Retrieving gene functional networks from knowledge databases presents a challenge due to the mismatch between disease networks and subtype-specific variations. Current solutions, including statistical and deep learning methods, often fail to effectively integrate gene interaction knowledge from databases or explicitly learn subtype-specific interactions. To address this mismatch, we propose GeSubNet, which learns a unified representation capable of predicting gene interactions while distinguishing between different disease subtypes. Graphs generated by such representations can be considered subtype-specific networks. GeSubNet is a multi-step representation learning framework with three modules: First, a deep generative model learns distinct disease subtypes from patient gene expression profiles. Second, a graph neural network captures representations of prior gene networks from knowledge databases, ensuring accurate physical gene interactions. Finally, we integrate these two representations using an inference loss that leverages graph generation capabilities, conditioned on the patient separation loss, to refine subtype-specific information in the learned representation. GeSubNet consistently outperforms traditional methods, with average improvements of 30.6%, 21.0%, 20.1%, and 56.6% across four graph evaluation metrics, averaged over four cancer datasets. Particularly, we conduct a biological simulation experiment to assess how the behavior of selected genes from over 11,000 candidates affects subtypes or patient distributions. The results show that the generated network has the potential to identify subtype-specific genes with an 83% likelihood of impacting patient distribution shifts. The GeSubNet resource is available: https://anonymous.4open.science/r/GeSubNet/
SplitSEE: A Splittable Self-supervised Framework for Single-Channel EEG Representation Learning
Oct 15, 2024While end-to-end multi-channel electroencephalography (EEG) learning approaches have shown significant promise, their applicability is often constrained in neurological diagnostics, such as intracranial EEG resources. When provided with a single-channel EEG, how can we learn representations that are robust to multi-channels and scalable across varied tasks, such as seizure prediction? In this paper, we present SplitSEE, a structurally splittable framework designed for effective temporal-frequency representation learning in single-channel EEG. The key concept of SplitSEE is a self-supervised framework incorporating a deep clustering task. Given an EEG, we argue that the time and frequency domains are two distinct perspectives, and hence, learned representations should share the same cluster assignment. To this end, we first propose two domain-specific modules that independently learn domain-specific representation and address the temporal-frequency tradeoff issue in conventional spectrogram-based methods. Then, we introduce a novel clustering loss to measure the information similarity. This encourages representations from both domains to coherently describe the same input by assigning them a consistent cluster. SplitSEE leverages a pre-training-to-fine-tuning framework within a splittable architecture and has following properties: (a) Effectiveness: it learns representations solely from single-channel EEG but has even outperformed multi-channel baselines. (b) Robustness: it shows the capacity to adapt across different channels with low performance variance. Superior performance is also achieved with our collected clinical dataset. (c) Scalability: With just one fine-tuning epoch, SplitSEE achieves high and stable performance using partial model layers.
CMOB: Large-Scale Cancer Multi-Omics Benchmark with Open Datasets, Tasks, and Baselines
Sep 02, 2024Machine learning has shown great potential in the field of cancer multi-omics studies, offering incredible opportunities for advancing precision medicine. However, the challenges associated with dataset curation and task formulation pose significant hurdles, especially for researchers lacking a biomedical background. Here, we introduce the CMOB, the first large-scale cancer multi-omics benchmark integrates the TCGA platform, making data resources accessible and usable for machine learning researchers without significant preparation and expertise.To date, CMOB includes a collection of 20 cancer multi-omics datasets covering 32 cancers, accompanied by a systematic data processing pipeline. CMOB provides well-processed dataset versions to support 20 meaningful tasks in four studies, with a collection of benchmarks. We also integrate CMOB with two complementary resources and various biological tools to explore broader research avenues.All resources are open-accessible with user-friendly and compatible integration scripts that enable non-experts to easily incorporate this complementary information for various tasks. We conduct extensive experiments on selected datasets to offer recommendations on suitable machine learning baselines for specific applications. Through CMOB, we aim to facilitate algorithmic advances and hasten the development, validation, and clinical translation of machine-learning models for personalized cancer treatments. CMOB is available on GitHub (\url{https://github.com/chenzRG/Cancer-Multi-Omics-Benchmark}).