 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Aspect Mining and Anomaly Detection for Heterogeneous Tensor Streams
Feb 04, 2026Analysis and anomaly detection in event tensor streams consisting of timestamps and multiple attributes - such as communication logs(time, IP address, packet length)- are essential tasks in data mining. While existing tensor decomposition and anomaly detection methods provide useful insights, they face the following two limitations. (i) They cannot handle heterogeneous tensor streams, which comprises both categorical attributes(e.g., IP address) and continuous attributes(e.g., packet length). They typically require either discretizing continuous attributes or treating categorical attributes as continuous, both of which distort the underlying statistical properties of the data.Furthermore, incorrect assumptions about the distribution family of continuous attributes often degrade the model's performance. (ii) They discretize timestamps, failing to track the temporal dynamics of streams(e.g., trends, abnormal events), which makes them ineffective for detecting anomalies at the group level, referred to as 'group anomalies' (e.g, DoS attacks). To address these challenges, we propose HeteroComp, a method for continuously summarizing heterogeneous tensor streams into 'components' representing latent groups in each attribute and their temporal dynamics, and detecting group anomalies. Our method employs Gaussian process priors to model unknown distributions of continuous attributes, and temporal dynamics, which directly estimate probability densities from data. Extracted components give concise but effective summarization, enabling accurate group anomaly detection. Extensive experiments on real datasets demonstrate that HeteroComp outperforms the state-of-the-art algorithms for group anomaly detection accuracy, and its computational time does not depend on the data stream length.
Fast Mining and Dynamic Time-to-Event Prediction over Multi-sensor Data Streams
Jan 08, 2026Given real-time sensor data streams obtained from machines, how can we continuously predict when a machine failure will occur? This work aims to continuously forecast the timing of future events by analyzing multi-sensor data streams. A key characteristic of real-world data streams is their dynamic nature, where the underlying patterns evolve over time. To address this, we present TimeCast, a dynamic prediction framework designed to adapt to these changes and provide accurate, real-time predictions of future event time. Our proposed method has the following properties: (a) Dynamic: it identifies the distinct time-evolving patterns (i.e., stages) and learns individual models for each, enabling us to make adaptive predictions based on pattern shifts. (b) Practical: it finds meaningful stages that capture time-varying interdependencies between multiple sensors and improve prediction performance; (c) Scalable: our algorithm scales linearly with the input size and enables online model updates on data streams. Extensive experiments on real datasets demonstrate that TimeCast provides higher prediction accuracy than state-of-the-art methods while finding dynamic changes in data streams with a great reduction in computational time.
EvoBrain: Dynamic Multi-channel EEG Graph Modeling for Time-evolving Brain Network
Sep 19, 2025Dynamic GNNs, which integrate temporal and spatial features in Electroencephalography (EEG) data, have shown great potential in automating seizure detection. However, fully capturing the underlying dynamics necessary to represent brain states, such as seizure and non-seizure, remains a non-trivial task and presents two fundamental challenges. First, most existing dynamic GNN methods are built on temporally fixed static graphs, which fail to reflect the evolving nature of brain connectivity during seizure progression. Second, current efforts to jointly model temporal signals and graph structures and, more importantly, their interactions remain nascent, often resulting in inconsistent performance. To address these challenges, we present the first theoretical analysis of these two problems, demonstrating the effectiveness and necessity of explicit dynamic modeling and time-then-graph dynamic GNN method. Building on these insights, we propose EvoBrain, a novel seizure detection model that integrates a two-stream Mamba architecture with a GCN enhanced by Laplacian Positional Encoding, following neurological insights. Moreover, EvoBrain incorporates explicitly dynamic graph structures, allowing both nodes and edges to evolve over time. Our contributions include (a) a theoretical analysis proving the expressivity advantage of explicit dynamic modeling and time-then-graph over other approaches, (b) a novel and efficient model that significantly improves AUROC by 23% and F1 score by 30%, compared with the dynamic GNN baseline, and (c) broad evaluations of our method on the challenging early seizure prediction tasks.
Robust and Explainable Detector of Time Series Anomaly via Augmenting Multiclass Pseudo-Anomalies
May 27, 2025Unsupervised anomaly detection in time series has been a pivotal research area for decades. Current mainstream approaches focus on learning normality, on the assumption that all or most of the samples in the training set are normal. However, anomalies in the training set (i.e., anomaly contamination) can be misleading. Recent studies employ data augmentation to generate pseudo-anomalies and learn the boundary separating the training samples from the augmented samples. Although this approach mitigates anomaly contamination if augmented samples mimic unseen real anomalies, it suffers from several limitations. (1) Covering a wide range of time series anomalies is challenging. (2) It disregards augmented samples that resemble normal samples (i.e., false anomalies). (3) It places too much trust in the labels of training and augmented samples. In response, we propose RedLamp, which employs diverse data augmentations to generate multiclass pseudo-anomalies and learns the multiclass boundary. Such multiclass pseudo-anomalies cover a wide variety of time series anomalies. We conduct multiclass classification using soft labels, which prevents the model from being overconfident and ensures its robustness against contaminated/false anomalies. The learned latent space is inherently explainable as it is trained to separate pseudo-anomalies into multiclasses. Extensive experiments demonstrate the effectiveness of RedLamp in anomaly detection and its robustness against anomaly contamination.
D-Tracker: Modeling Interest Diffusion in Social Activity Tensor Data Streams
May 01, 2025Large quantities of social activity data, such as weekly web search volumes and the number of new infections with infectious diseases, reflect peoples' interests and activities. It is important to discover temporal patterns from such data and to forecast future activities accurately. However, modeling and forecasting social activity data streams is difficult because they are high-dimensional and composed of multiple time-varying dynamics such as trends, seasonality, and interest diffusion. In this paper, we propose D-Tracker, a method for continuously capturing time-varying temporal patterns within social activity tensor data streams and forecasting future activities. Our proposed method has the following properties: (a) Interpretable: it incorporates the partial differential equation into a tensor decomposition framework and captures time-varying temporal patterns such as trends, seasonality, and interest diffusion between locations in an interpretable manner; (b) Automatic: it has no hyperparameters and continuously models tensor data streams fully automatically; (c) Scalable: the computation time of D-Tracker is independent of the time series length. Experiments using web search volume data obtained from GoogleTrends, and COVID-19 infection data obtained from COVID-19 Open Data Repository show that our method can achieve higher forecasting accuracy in less computation time than existing methods while extracting the interest diffusion between locations. Our source code and datasets are available at {https://github.com/Higashiguchi-Shingo/D-Tracker.
ExPath: Towards Explaining Targeted Pathways for Biological Knowledge Bases
Feb 25, 2025Biological knowledge bases provide systemically functional pathways of cells or organisms in terms of molecular interaction. However, recognizing more targeted pathways, particularly when incorporating wet-lab experimental data, remains challenging and typically requires downstream biological analyses and expertise. In this paper, we frame this challenge as a solvable graph learning and explaining task and propose a novel pathway inference framework, ExPath, that explicitly integrates experimental data, specifically amino acid sequences (AA-seqs), to classify various graphs (bio-networks) in biological databases. The links (representing pathways) that contribute more to classification can be considered as targeted pathways. Technically, ExPath comprises three components: (1) a large protein language model (pLM) that encodes and embeds AA-seqs into graph, overcoming traditional obstacles in processing AA-seq data, such as BLAST; (2) PathMamba, a hybrid architecture combining graph neural networks (GNNs) with state-space sequence modeling (Mamba) to capture both local interactions and global pathway-level dependencies; and (3) PathExplainer, a subgraph learning module that identifies functionally critical nodes and edges through trainable pathway masks. We also propose ML-oriented biological evaluations and a new metric. The experiments involving 301 bio-networks evaluations demonstrate that pathways inferred by ExPath maintain biological meaningfulness. We will publicly release curated 301 bio-network data soon.
Modeling Time-evolving Causality over Data Streams
Feb 13, 2025Given an extensive, semi-infinite collection of multivariate coevolving data sequences (e.g., sensor/web activity streams) whose observations influence each other, how can we discover the time-changing cause-and-effect relationships in co-evolving data streams? How efficiently can we reveal dynamical patterns that allow us to forecast future values? In this paper, we present a novel streaming method, ModePlait, which is designed for modeling such causal relationships (i.e., time-evolving causality) in multivariate co-evolving data streams and forecasting their future values. The solution relies on characteristics of the causal relationships that evolve over time in accordance with the dynamic changes of exogenous variables. ModePlait has the following properties: (a) Effective: it discovers the time-evolving causality in multivariate co-evolving data streams by detecting the transitions of distinct dynamical patterns adaptively. (b) Accurate: it enables both the discovery of time-evolving causality and the forecasting of future values in a streaming fashion. (c) Scalable: our algorithm does not depend on data stream length and thus is applicable to very large sequences. Extensive experiments on both synthetic and real-world datasets demonstrate that our proposed model outperforms state-of-the-art methods in terms of discovering the time-evolving causality as well as forecasting.
SODor: Long-Term EEG Partitioning for Seizure Onset Detection
Dec 20, 2024



Deep learning models have recently shown great success in classifying epileptic patients using EEG recordings. Unfortunately, classification-based methods lack a sound mechanism to detect the onset of seizure events. In this work, we propose a two-stage framework, \method, that explicitly models seizure onset through a novel task formulation of subsequence clustering. Given an EEG sequence, the framework first learns a set of second-level embeddings with label supervision. It then employs model-based clustering to explicitly capture long-term temporal dependencies in EEG sequences and identify meaningful subsequences. Epochs within a subsequence share a common cluster assignment (normal or seizure), with cluster or state transitions representing successful onset detections. Extensive experiments on three datasets demonstrate that our method can correct misclassifications, achieving 5%-11% classification improvements over other baselines and accurately detecting seizure onsets.
Modeling Latent Non-Linear Dynamical System over Time Series
Dec 12, 2024We study the problem of modeling a non-linear dynamical system when given a time series by deriving equations directly from the data. Despite the fact that time series data are given as input, models for dynamics and estimation algorithms that incorporate long-term temporal dependencies are largely absent from existing studies. In this paper, we introduce a latent state to allow time-dependent modeling and formulate this problem as a dynamics estimation problem in latent states. We face multiple technical challenges, including (1) modeling latent non-linear dynamics and (2) solving circular dependencies caused by the presence of latent states. To tackle these challenging problems, we propose a new method, Latent Non-Linear equation modeling (LaNoLem), that can model a latent non-linear dynamical system and a novel alternating minimization algorithm for effectively estimating latent states and model parameters. In addition, we introduce criteria to control model complexity without human intervention. Compared with the state-of-the-art model, LaNoLem achieves competitive performance for estimating dynamics while outperforming other methods in prediction.
GeSubNet: Gene Interaction Inference for Disease Subtype Network Generation
Oct 17, 2024


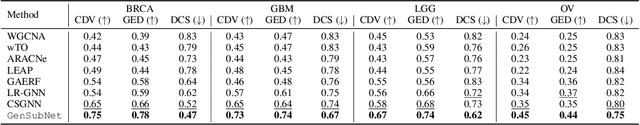
Retrieving gene functional networks from knowledge databases presents a challenge due to the mismatch between disease networks and subtype-specific variations. Current solutions, including statistical and deep learning methods, often fail to effectively integrate gene interaction knowledge from databases or explicitly learn subtype-specific interactions. To address this mismatch, we propose GeSubNet, which learns a unified representation capable of predicting gene interactions while distinguishing between different disease subtypes. Graphs generated by such representations can be considered subtype-specific networks. GeSubNet is a multi-step representation learning framework with three modules: First, a deep generative model learns distinct disease subtypes from patient gene expression profiles. Second, a graph neural network captures representations of prior gene networks from knowledge databases, ensuring accurate physical gene interactions. Finally, we integrate these two representations using an inference loss that leverages graph generation capabilities, conditioned on the patient separation loss, to refine subtype-specific information in the learned representation. GeSubNet consistently outperforms traditional methods, with average improvements of 30.6%, 21.0%, 20.1%, and 56.6% across four graph evaluation metrics, averaged over four cancer datasets. Particularly, we conduct a biological simulation experiment to assess how the behavior of selected genes from over 11,000 candidates affects subtypes or patient distributions. The results show that the generated network has the potential to identify subtype-specific genes with an 83% likelihood of impacting patient distribution shifts. The GeSubNet resource is available: https://anonymous.4open.science/r/GeSubNet/