 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCategory Prompt Mamba Network for Nuclei Segmentation and Classification
Mar 13, 2025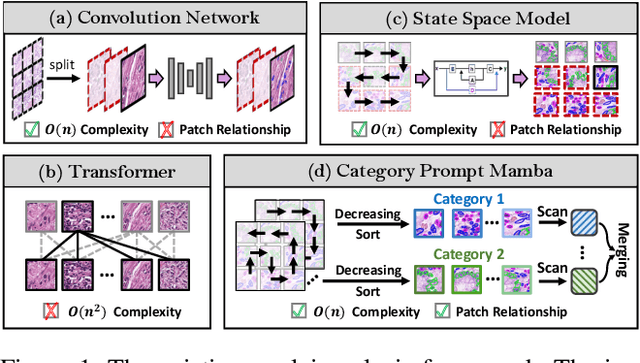
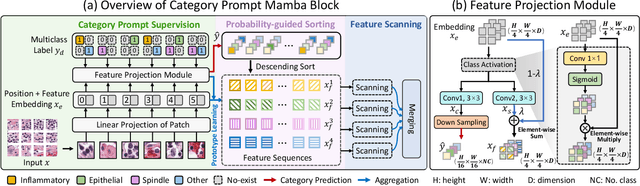
Nuclei segmentation and classification provide an essential basis for tumor immune microenvironment analysis. The previous nuclei segmentation and classification models require splitting large images into smaller patches for training, leading to two significant issues. First, nuclei at the borders of adjacent patches often misalign during inference. Second, this patch-based approach significantly increases the model's training and inference time. Recently, Mamba has garnered attention for its ability to model large-scale images with linear time complexity and low memory consumption. It offers a promising solution for training nuclei segmentation and classification models on full-sized images. However, the Mamba orientation-based scanning method lacks account for category-specific features, resulting in sub-optimal performance in scenarios with imbalanced class distributions. To address these challenges, this paper introduces a novel scanning strategy based on category probability sorting, which independently ranks and scans features for each category according to confidence from high to low. This approach enhances the feature representation of uncertain samples and mitigates the issues caused by imbalanced distributions. Extensive experiments conducted on four public datasets demonstrate that our method outperforms state-of-the-art approaches, delivering superior performance in nuclei segmentation and classification tasks.
HisynSeg: Weakly-Supervised Histopathological Image Segmentation via Image-Mixing Synthesis and Consistency Regularization
Dec 30, 2024



Tissue semantic segmentation is one of the key tasks in computational pathology. To avoid the expensive and laborious acquisition of pixel-level annotations, a wide range of studies attempt to adopt the class activation map (CAM), a weakly-supervised learning scheme, to achieve pixel-level tissue segmentation. However, CAM-based methods are prone to suffer from under-activation and over-activation issues, leading to poor segmentation performance. To address this problem, we propose a novel weakly-supervised semantic segmentation framework for histopathological images based on image-mixing synthesis and consistency regularization, dubbed HisynSeg. Specifically, synthesized histopathological images with pixel-level masks are generated for fully-supervised model training, where two synthesis strategies are proposed based on Mosaic transformation and B\'ezier mask generation. Besides, an image filtering module is developed to guarantee the authenticity of the synthesized images. In order to further avoid the model overfitting to the occasional synthesis artifacts, we additionally propose a novel self-supervised consistency regularization, which enables the real images without segmentation masks to supervise the training of the segmentation model. By integrating the proposed techniques, the HisynSeg framework successfully transforms the weakly-supervised semantic segmentation problem into a fully-supervised one, greatly improving the segmentation accuracy. Experimental results on three datasets prove that the proposed method achieves a state-of-the-art performance. Code is available at https://github.com/Vison307/HisynSeg.
DAWN: Domain-Adaptive Weakly Supervised Nuclei Segmentation via Cross-Task Interactions
Apr 24, 2024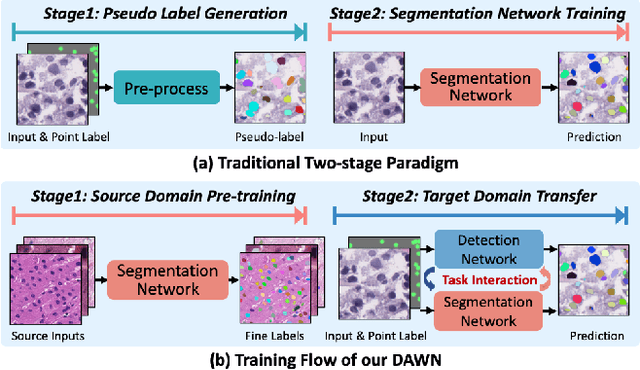
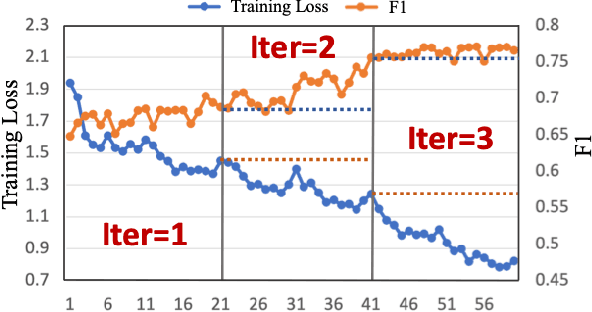

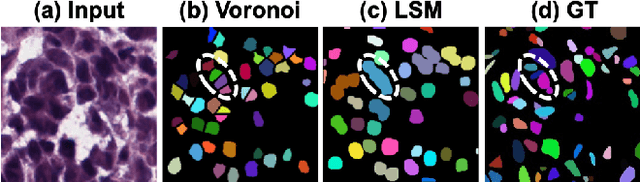
Weakly supervised segmentation methods have gained significant attention due to their ability to reduce the reliance on costly pixel-level annotations during model training. However, the current weakly supervised nuclei segmentation approaches typically follow a two-stage pseudo-label generation and network training process. The performance of the nuclei segmentation heavily relies on the quality of the generated pseudo-labels, thereby limiting its effectiveness. This paper introduces a novel domain-adaptive weakly supervised nuclei segmentation framework using cross-task interaction strategies to overcome the challenge of pseudo-label generation. Specifically, we utilize weakly annotated data to train an auxiliary detection task, which assists the domain adaptation of the segmentation network. To enhance the efficiency of domain adaptation, we design a consistent feature constraint module integrating prior knowledge from the source domain. Furthermore, we develop pseudo-label optimization and interactive training methods to improve the domain transfer capability. To validate the effectiveness of our proposed method, we conduct extensive comparative and ablation experiments on six datasets. The results demonstrate the superiority of our approach over existing weakly supervised approaches. Remarkably, our method achieves comparable or even better performance than fully supervised methods. Our code will be released in https://github.com/zhangye-zoe/DAWN.
AV-GAN: Attention-Based Varifocal Generative Adversarial Network for Uneven Medical Image Translation
Apr 16, 2024Different types of staining highlight different structures in organs, thereby assisting in diagnosis. However, due to the impossibility of repeated staining, we cannot obtain different types of stained slides of the same tissue area. Translating the slide that is easy to obtain (e.g., H&E) to slides of staining types difficult to obtain (e.g., MT, PAS) is a promising way to solve this problem. However, some regions are closely connected to other regions, and to maintain this connection, they often have complex structures and are difficult to translate, which may lead to wrong translations. In this paper, we propose the Attention-Based Varifocal Generative Adversarial Network (AV-GAN), which solves multiple problems in pathologic image translation tasks, such as uneven translation difficulty in different regions, mutual interference of multiple resolution information, and nuclear deformation. Specifically, we develop an Attention-Based Key Region Selection Module, which can attend to regions with higher translation difficulty. We then develop a Varifocal Module to translate these regions at multiple resolutions. Experimental results show that our proposed AV-GAN outperforms existing image translation methods with two virtual kidney tissue staining tasks and improves FID values by 15.9 and 4.16 respectively in the H&E-MT and H&E-PAS tasks.
MamMIL: Multiple Instance Learning for Whole Slide Images with State Space Models
Mar 08, 2024



Recently, pathological diagnosis, the gold standard for cancer diagnosis, has achieved superior performance by combining the Transformer with the multiple instance learning (MIL) framework using whole slide images (WSIs). However, the giga-pixel nature of WSIs poses a great challenge for the quadratic-complexity self-attention mechanism in Transformer to be applied in MIL. Existing studies usually use linear attention to improve computing efficiency but inevitably bring performance bottlenecks. To tackle this challenge, we propose a MamMIL framework for WSI classification by cooperating the selective structured state space model (i.e., Mamba) with MIL for the first time, enabling the modeling of instance dependencies while maintaining linear complexity. Specifically, to solve the problem that Mamba can only conduct unidirectional one-dimensional (1D) sequence modeling, we innovatively introduce a bidirectional state space model and a 2D context-aware block to enable MamMIL to learn the bidirectional instance dependencies with 2D spatial relationships. Experiments on two datasets show that MamMIL can achieve advanced classification performance with smaller memory footprints than the state-of-the-art MIL frameworks based on the Transformer. The code will be open-sourced if accepted.
Deep Learning Predicts Biomarker Status and Discovers Related Histomorphology Characteristics for Low-Grade Glioma
Oct 11, 2023



Biomarker detection is an indispensable part in the diagnosis and treatment of low-grade glioma (LGG). However, current LGG biomarker detection methods rely on expensive and complex molecular genetic testing, for which professionals are required to analyze the results, and intra-rater variability is often reported. To overcome these challenges, we propose an interpretable deep learning pipeline, a Multi-Biomarker Histomorphology Discoverer (Multi-Beholder) model based on the multiple instance learning (MIL) framework, to predict the status of five biomarkers in LGG using only hematoxylin and eosin-stained whole slide images and slide-level biomarker status labels. Specifically, by incorporating the one-class classification into the MIL framework, accurate instance pseudo-labeling is realized for instance-level supervision, which greatly complements the slide-level labels and improves the biomarker prediction performance. Multi-Beholder demonstrates superior prediction performance and generalizability for five LGG biomarkers (AUROC=0.6469-0.9735) in two cohorts (n=607) with diverse races and scanning protocols. Moreover, the excellent interpretability of Multi-Beholder allows for discovering the quantitative and qualitative correlations between biomarker status and histomorphology characteristics. Our pipeline not only provides a novel approach for biomarker prediction, enhancing the applicability of molecular treatments for LGG patients but also facilitates the discovery of new mechanisms in molecular functionality and LGG progression.
CoNIC Challenge: Pushing the Frontiers of Nuclear Detection, Segmentation, Classification and Counting
Mar 14, 2023



Nuclear detection, segmentation and morphometric profiling are essential in helping us further understand the relationship between histology and patient outcome. To drive innovation in this area, we setup a community-wide challenge using the largest available dataset of its kind to assess nuclear segmentation and cellular composition. Our challenge, named CoNIC, stimulated the development of reproducible algorithms for cellular recognition with real-time result inspection on public leaderboards. We conducted an extensive post-challenge analysis based on the top-performing models using 1,658 whole-slide images of colon tissue. With around 700 million detected nuclei per model, associated features were used for dysplasia grading and survival analysis, where we demonstrated that the challenge's improvement over the previous state-of-the-art led to significant boosts in downstream performance. Our findings also suggest that eosinophils and neutrophils play an important role in the tumour microevironment. We release challenge models and WSI-level results to foster the development of further methods for biomarker discovery.
WSSS4LUAD: Grand Challenge on Weakly-supervised Tissue Semantic Segmentation for Lung Adenocarcinoma
Apr 14, 2022
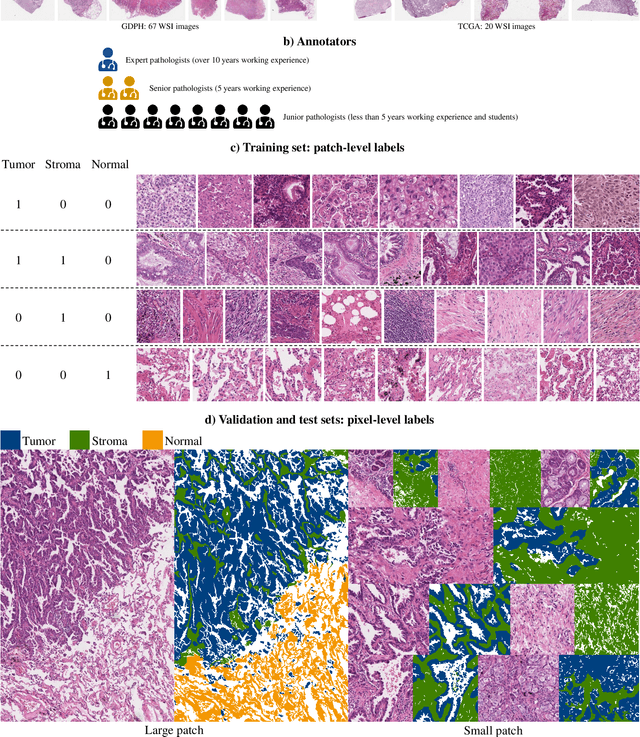
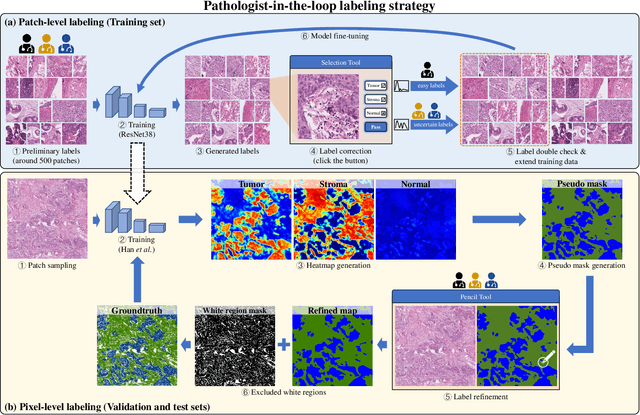
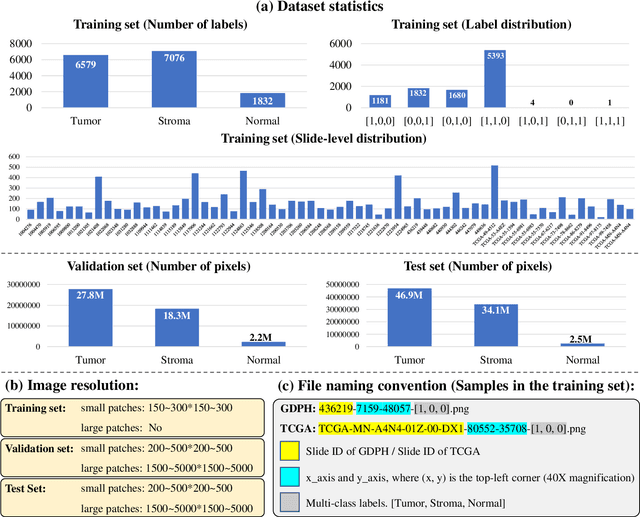
Lung cancer is the leading cause of cancer death worldwide, and adenocarcinoma (LUAD) is the most common subtype. Exploiting the potential value of the histopathology images can promote precision medicine in oncology. Tissue segmentation is the basic upstream task of histopathology image analysis. Existing deep learning models have achieved superior segmentation performance but require sufficient pixel-level annotations, which is time-consuming and expensive. To enrich the label resources of LUAD and to alleviate the annotation efforts, we organize this challenge WSSS4LUAD to call for the outstanding weakly-supervised semantic segmentation (WSSS) techniques for histopathology images of LUAD. Participants have to design the algorithm to segment tumor epithelial, tumor-associated stroma and normal tissue with only patch-level labels. This challenge includes 10,091 patch-level annotations (the training set) and over 130 million labeled pixels (the validation and test sets), from 87 WSIs (67 from GDPH, 20 from TCGA). All the labels were generated by a pathologist-in-the-loop pipeline with the help of AI models and checked by the label review board. Among 532 registrations, 28 teams submitted the results in the test phase with over 1,000 submissions. Finally, the first place team achieved mIoU of 0.8413 (tumor: 0.8389, stroma: 0.7931, normal: 0.8919). According to the technical reports of the top-tier teams, CAM is still the most popular approach in WSSS. Cutmix data augmentation has been widely adopted to generate more reliable samples. With the success of this challenge, we believe that WSSS approaches with patch-level annotations can be a complement to the traditional pixel annotations while reducing the annotation efforts. The entire dataset has been released to encourage more researches on computational pathology in LUAD and more novel WSSS techniques.