 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePhenotype discovery of traumatic brain injury segmentations from heterogeneous multi-site data
Nov 05, 2025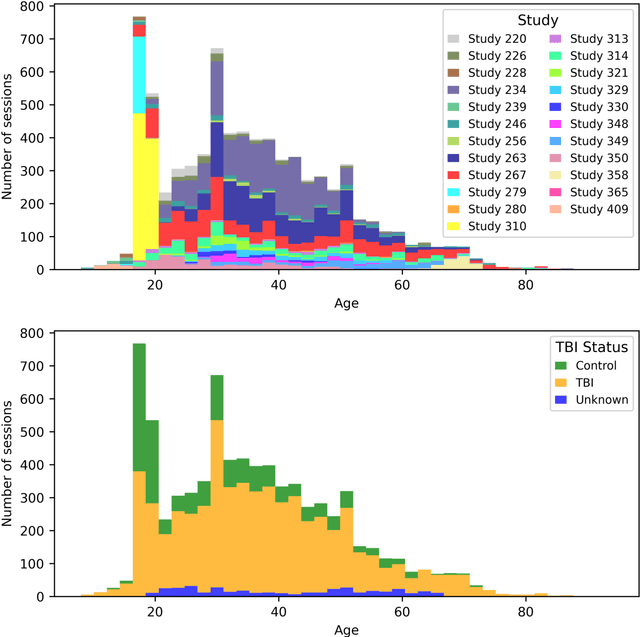
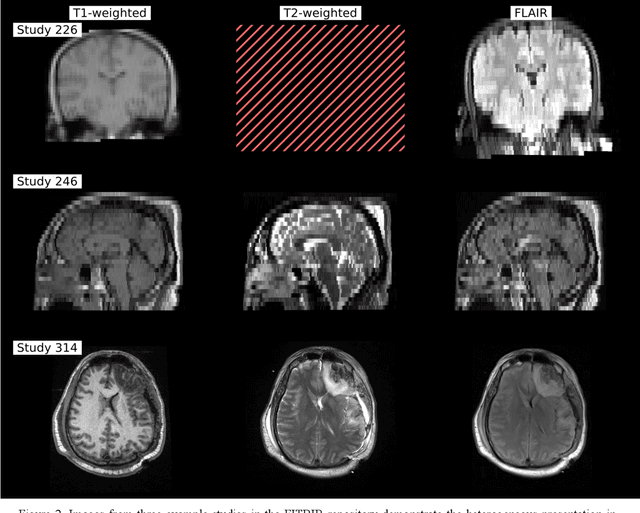
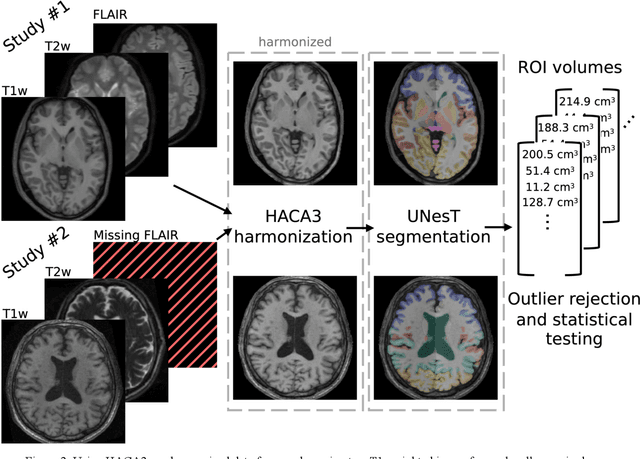
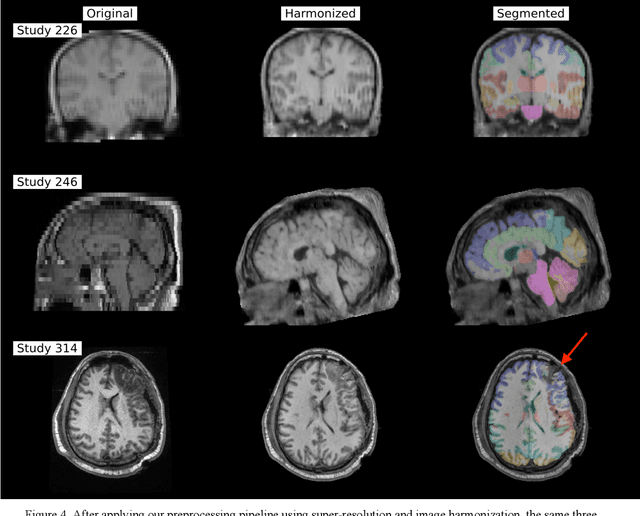
Traumatic brain injury (TBI) is intrinsically heterogeneous, and typical clinical outcome measures like the Glasgow Coma Scale complicate this diversity. The large variability in severity and patient outcomes render it difficult to link structural damage to functional deficits. The Federal Interagency Traumatic Brain Injury Research (FITBIR) repository contains large-scale multi-site magnetic resonance imaging data of varying resolutions and acquisition parameters (25 shared studies with 7,693 sessions that have age, sex and TBI status defined - 5,811 TBI and 1,882 controls). To reveal shared pathways of injury of TBI through imaging, we analyzed T1-weighted images from these sessions by first harmonizing to a local dataset and segmenting 132 regions of interest (ROIs) in the brain. After running quality assurance, calculating the volumes of the ROIs, and removing outliers, we calculated the z-scores of volumes for all participants relative to the mean and standard deviation of the controls. We regressed out sex, age, and total brain volume with a multivariate linear regression, and we found significant differences in 37 ROIs between subjects with TBI and controls (p < 0.05 with independent t-tests with false discovery rate correction). We found that differences originated in 1) the brainstem, occipital pole and structures posterior to the orbit, 2) subcortical gray matter and insular cortex, and 3) cerebral and cerebellar white matter using independent component analysis and clustering the component loadings of those with TBI.
DeepFixel: Crossing white matter fiber identification through spherical convolutional neural networks
Nov 05, 2025Diffusion-weighted magnetic resonance imaging allows for reconstruction of models for structural connectivity in the brain, such as fiber orientation distribution functions (ODFs) that describe the distribution, direction, and volume of white matter fiber bundles in a voxel. Crossing white matter fibers in voxels complicate analysis and can lead to errors in downstream tasks like tractography. We introduce one option for separating fiber ODFs by performing a nonlinear optimization to fit ODFs to the given data and penalizing terms that are not symmetric about the axis of the fiber. However, this optimization is non-convex and computationally infeasible across an entire image (approximately 1.01 x 106 ms per voxel). We introduce DeepFixel, a spherical convolutional neural network approximation for this nonlinear optimization. We model the probability distribution of fibers as a spherical mesh with higher angular resolution than a truncated spherical harmonic representation. To validate DeepFixel, we compare to the nonlinear optimization and a fixel-based separation algorithm of two-fiber and three-fiber ODFs. The median angular correlation coefficient is 1 (interquartile range of 0.00) using the nonlinear optimization algorithm, 0.988 (0.317) using a fiber bundle elements or "fixel"-based separation algorithm, and 0.973 (0.004) using DeepFixel. DeepFixel is more computationally efficient than the non-convex optimization (0.32 ms per voxel). DeepFixel's spherical mesh representation is successful at disentangling at smaller angular separations and smaller volume fractions than the fixel-based separation algorithm.
Lifespan Pancreas Morphology for Control vs Type 2 Diabetes using AI on Largescale Clinical Imaging
Aug 20, 2025Purpose: Understanding how the pancreas changes is critical for detecting deviations in type 2 diabetes and other pancreatic disease. We measure pancreas size and shape using morphological measurements from ages 0 to 90. Our goals are to 1) identify reliable clinical imaging modalities for AI-based pancreas measurement, 2) establish normative morphological aging trends, and 3) detect potential deviations in type 2 diabetes. Approach: We analyzed a clinically acquired dataset of 2533 patients imaged with abdominal CT or MRI. We resampled the scans to 3mm isotropic resolution, segmented the pancreas using automated methods, and extracted 13 morphological pancreas features across the lifespan. First, we assessed CT and MRI measurements to determine which modalities provide consistent lifespan trends. Second, we characterized distributions of normative morphological patterns stratified by age group and sex. Third, we used GAMLSS regression to model pancreas morphology trends in 1350 patients matched for age, sex, and type 2 diabetes status to identify any deviations from normative aging associated with type 2 diabetes. Results: When adjusting for confounders, the aging trends for 10 of 13 morphological features were significantly different between patients with type 2 diabetes and non-diabetic controls (p < 0.05 after multiple comparisons corrections). Additionally, MRI appeared to yield different pancreas measurements than CT using our AI-based method. Conclusions: We provide lifespan trends demonstrating that the size and shape of the pancreas is altered in type 2 diabetes using 675 control patients and 675 diabetes patients. Moreover, our findings reinforce that the pancreas is smaller in type 2 diabetes. Additionally, we contribute a reference of lifespan pancreas morphology from a large cohort of non-diabetic control patients in a clinical setting.
Multipath cycleGAN for harmonization of paired and unpaired low-dose lung computed tomography reconstruction kernels
May 28, 2025Reconstruction kernels in computed tomography (CT) affect spatial resolution and noise characteristics, introducing systematic variability in quantitative imaging measurements such as emphysema quantification. Choosing an appropriate kernel is therefore essential for consistent quantitative analysis. We propose a multipath cycleGAN model for CT kernel harmonization, trained on a mixture of paired and unpaired data from a low-dose lung cancer screening cohort. The model features domain-specific encoders and decoders with a shared latent space and uses discriminators tailored for each domain.We train the model on 42 kernel combinations using 100 scans each from seven representative kernels in the National Lung Screening Trial (NLST) dataset. To evaluate performance, 240 scans from each kernel are harmonized to a reference soft kernel, and emphysema is quantified before and after harmonization. A general linear model assesses the impact of age, sex, smoking status, and kernel on emphysema. We also evaluate harmonization from soft kernels to a reference hard kernel. To assess anatomical consistency, we compare segmentations of lung vessels, muscle, and subcutaneous adipose tissue generated by TotalSegmentator between harmonized and original images. Our model is benchmarked against traditional and switchable cycleGANs. For paired kernels, our approach reduces bias in emphysema scores, as seen in Bland-Altman plots (p<0.05). For unpaired kernels, harmonization eliminates confounding differences in emphysema (p>0.05). High Dice scores confirm preservation of muscle and fat anatomy, while lung vessel overlap remains reasonable. Overall, our shared latent space multipath cycleGAN enables robust harmonization across paired and unpaired CT kernels, improving emphysema quantification and preserving anatomical fidelity.
Investigating the impact of kernel harmonization and deformable registration on inspiratory and expiratory chest CT images for people with COPD
Feb 07, 2025



Paired inspiratory-expiratory CT scans enable the quantification of gas trapping due to small airway disease and emphysema by analyzing lung tissue motion in COPD patients. Deformable image registration of these scans assesses regional lung volumetric changes. However, variations in reconstruction kernels between paired scans introduce errors in quantitative analysis. This work proposes a two-stage pipeline to harmonize reconstruction kernels and perform deformable image registration using data acquired from the COPDGene study. We use a cycle generative adversarial network (GAN) to harmonize inspiratory scans reconstructed with a hard kernel (BONE) to match expiratory scans reconstructed with a soft kernel (STANDARD). We then deformably register the expiratory scans to inspiratory scans. We validate harmonization by measuring emphysema using a publicly available segmentation algorithm before and after harmonization. Results show harmonization significantly reduces emphysema measurement inconsistencies, decreasing median emphysema scores from 10.479% to 3.039%, with a reference median score of 1.305% from the STANDARD kernel as the target. Registration accuracy is evaluated via Dice overlap between emphysema regions on inspiratory, expiratory, and deformed images. The Dice coefficient between inspiratory emphysema masks and deformably registered emphysema masks increases significantly across registration stages (p<0.001). Additionally, we demonstrate that deformable registration is robust to kernel variations.
Sensitivity of quantitative diffusion MRI tractography and microstructure to anisotropic spatial sampling
Sep 26, 2024Purpose: Diffusion weighted MRI (dMRI) and its models of neural structure provide insight into human brain organization and variations in white matter. A recent study by McMaster, et al. showed that complex graph measures of the connectome, the graphical representation of a tractogram, vary with spatial sampling changes, but biases introduced by anisotropic voxels in the process have not been well characterized. This study uses microstructural measures (fractional anisotropy and mean diffusivity) and white matter bundle properties (bundle volume, length, and surface area) to further understand the effect of anisotropic voxels on microstructure and tractography. Methods: The statistical significance of the selected measures derived from dMRI data were assessed by comparing three white matter bundles at different spatial resolutions with 44 subjects from the Human Connectome Project Young Adult dataset scan/rescan data using the Wilcoxon Signed Rank test. The original isotropic resolution (1.25 mm isotropic) was explored with six anisotropic resolutions with 0.25 mm incremental steps in the z dimension. Then, all generated resolutions were upsampled to 1.25 mm isotropic and 1 mm isotropic. Results: There were statistically significant differences between at least one microstructural and one bundle measure at every resolution (p less than or equal to 0.05, corrected for multiple comparisons). Cohen's d coefficient evaluated the effect size of anisotropic voxels on microstructure and tractography. Conclusion: Fractional anisotropy and mean diffusivity cannot be recovered with basic up sampling from low quality data with gold standard data. However, the bundle measures from tractogram become more repeatable when voxels are resampled to 1 mm isotropic.
Influence of Early through Late Fusion on Pancreas Segmentation from Imperfectly Registered Multimodal MRI
Sep 06, 2024Multimodal fusion promises better pancreas segmentation. However, where to perform fusion in models is still an open question. It is unclear if there is a best location to fuse information when analyzing pairs of imperfectly aligned images. Two main alignment challenges in this pancreas segmentation study are 1) the pancreas is deformable and 2) breathing deforms the abdomen. Even after image registration, relevant deformations are often not corrected. We examine how early through late fusion impacts pancreas segmentation. We used 353 pairs of T2-weighted (T2w) and T1-weighted (T1w) abdominal MR images from 163 subjects with accompanying pancreas labels. We used image registration (deeds) to align the image pairs. We trained a collection of basic UNets with different fusion points, spanning from early to late, to assess how early through late fusion influenced segmentation performance on imperfectly aligned images. We assessed generalization of fusion points on nnUNet. The single-modality T2w baseline using a basic UNet model had a Dice score of 0.73, while the same baseline on the nnUNet model achieved 0.80. For the basic UNet, the best fusion approach occurred in the middle of the encoder (early/mid fusion), which led to a statistically significant improvement of 0.0125 on Dice score compared to the baseline. For the nnUNet, the best fusion approach was na\"ive image concatenation before the model (early fusion), which resulted in a statistically significant Dice score increase of 0.0021 compared to baseline. Fusion in specific blocks can improve performance, but the best blocks for fusion are model specific, and the gains are small. In imperfectly registered datasets, fusion is a nuanced problem, with the art of design remaining vital for uncovering potential insights. Future innovation is needed to better address fusion in cases of imperfect alignment of abdominal image pairs.
Harmonized connectome resampling for variance in voxel sizes
Aug 02, 2024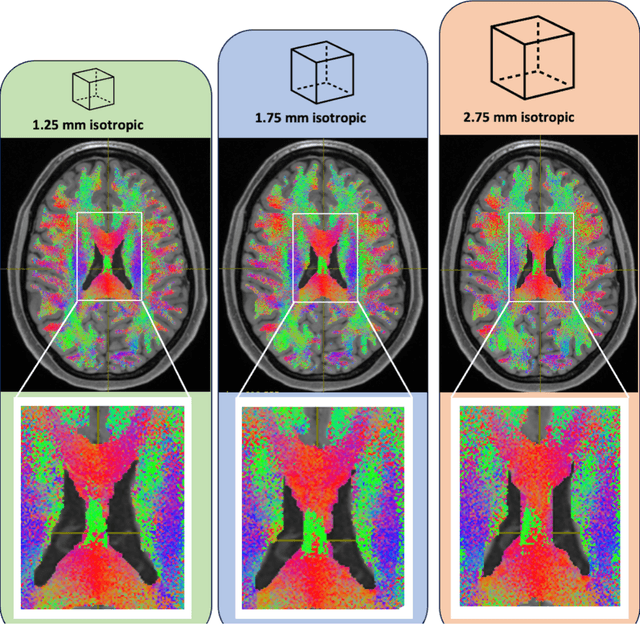
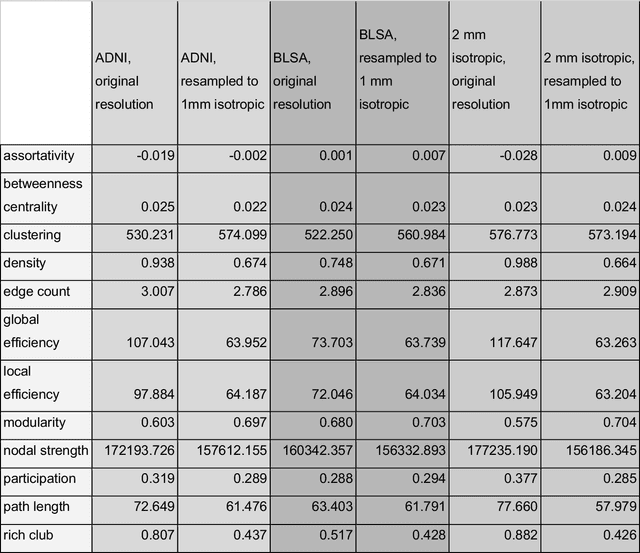
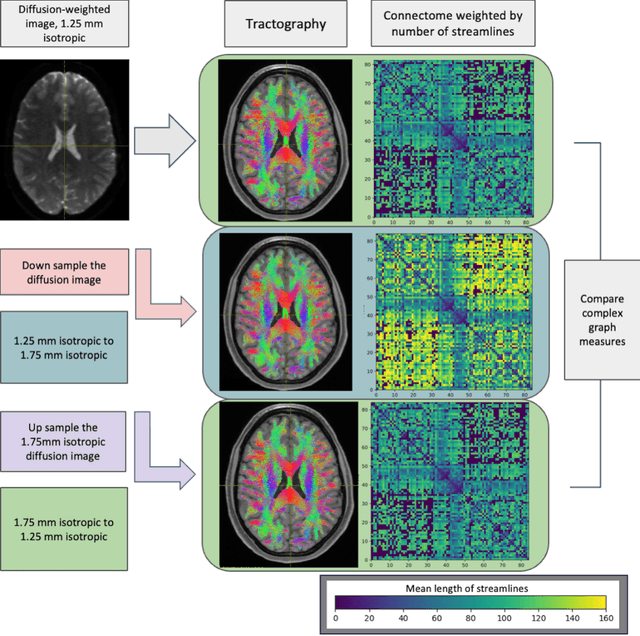
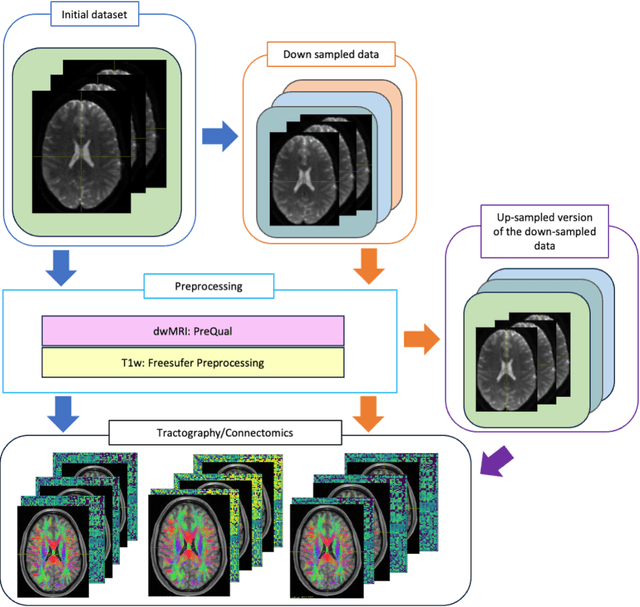
To date, there has been no comprehensive study characterizing the effect of diffusion-weighted magnetic resonance imaging voxel resolution on the resulting connectome for high resolution subject data. Similarity in results improved with higher resolution, even after initial down-sampling. To ensure robust tractography and connectomes, resample data to 1 mm isotropic resolution.
Enhancing Single-Slice Segmentation with 3D-to-2D Unpaired Scan Distillation
Jun 18, 2024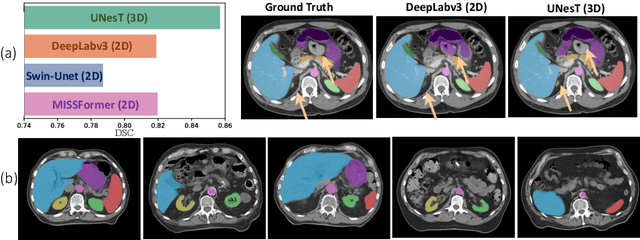
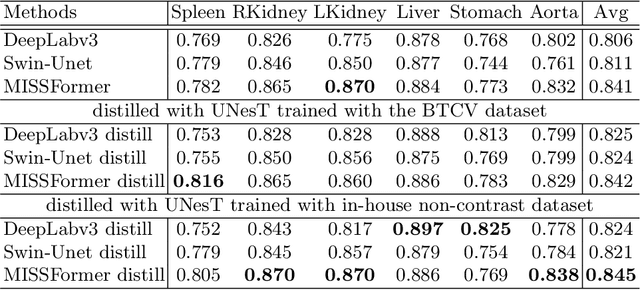
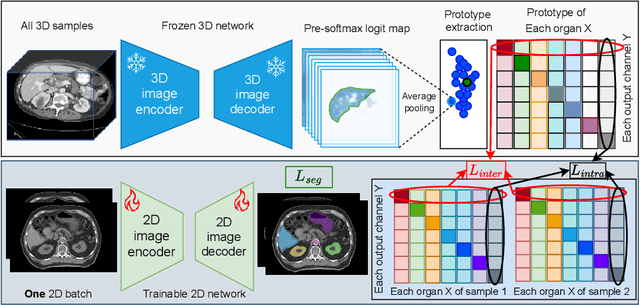
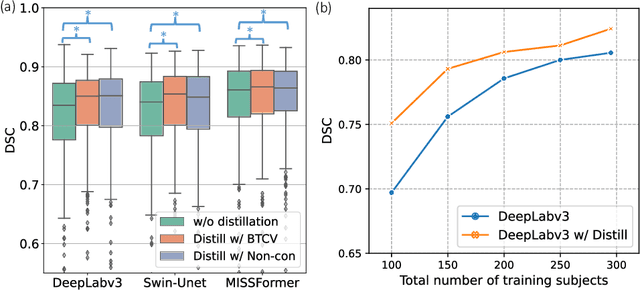
2D single-slice abdominal computed tomography (CT) enables the assessment of body habitus and organ health with low radiation exposure. However, single-slice data necessitates the use of 2D networks for segmentation, but these networks often struggle to capture contextual information effectively. Consequently, even when trained on identical datasets, 3D networks typically achieve superior segmentation results. In this work, we propose a novel 3D-to-2D distillation framework, leveraging pre-trained 3D models to enhance 2D single-slice segmentation. Specifically, we extract the prediction distribution centroid from the 3D representations, to guide the 2D student by learning intra- and inter-class correlation. Unlike traditional knowledge distillation methods that require the same data input, our approach employs unpaired 3D CT scans with any contrast to guide the 2D student model. Experiments conducted on 707 subjects from the single-slice Baltimore Longitudinal Study of Aging (BLSA) dataset demonstrate that state-of-the-art 2D multi-organ segmentation methods can benefit from the 3D teacher model, achieving enhanced performance in single-slice multi-organ segmentation. Notably, our approach demonstrates considerable efficacy in low-data regimes, outperforming the model trained with all available training subjects even when utilizing only 200 training subjects. Thus, this work underscores the potential to alleviate manual annotation burdens.
Nucleus subtype classification using inter-modality learning
Jan 29, 2024Understanding the way cells communicate, co-locate, and interrelate is essential to understanding human physiology. Hematoxylin and eosin (H&E) staining is ubiquitously available both for clinical studies and research. The Colon Nucleus Identification and Classification (CoNIC) Challenge has recently innovated on robust artificial intelligence labeling of six cell types on H&E stains of the colon. However, this is a very small fraction of the number of potential cell classification types. Specifically, the CoNIC Challenge is unable to classify epithelial subtypes (progenitor, endocrine, goblet), lymphocyte subtypes (B, helper T, cytotoxic T), or connective subtypes (fibroblasts, stromal). In this paper, we propose to use inter-modality learning to label previously un-labelable cell types on virtual H&E. We leveraged multiplexed immunofluorescence (MxIF) histology imaging to identify 14 subclasses of cell types. We performed style transfer to synthesize virtual H&E from MxIF and transferred the higher density labels from MxIF to these virtual H&E images. We then evaluated the efficacy of learning in this approach. We identified helper T and progenitor nuclei with positive predictive values of $0.34 \pm 0.15$ (prevalence $0.03 \pm 0.01$) and $0.47 \pm 0.1$ (prevalence $0.07 \pm 0.02$) respectively on virtual H&E. This approach represents a promising step towards automating annotation in digital pathology.