 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCausal ATE Mitigates Unintended Bias in Controlled Text Generation
Nov 19, 2023We study attribute control in language models through the method of Causal Average Treatment Effect (Causal ATE). Existing methods for the attribute control task in Language Models (LMs) check for the co-occurrence of words in a sentence with the attribute of interest, and control for them. However, spurious correlation of the words with the attribute in the training dataset, can cause models to hallucinate the presence of the attribute when presented with the spurious correlate during inference. We show that the simple perturbation-based method of Causal ATE removes this unintended effect. Additionally, we offer a theoretical foundation for investigating Causal ATE in the classification task, and prove that it reduces the number of false positives -- thereby mitigating the issue of unintended bias. Specifically, we ground it in the problem of toxicity mitigation, where a significant challenge lies in the inadvertent bias that often emerges towards protected groups post detoxification. We show that this unintended bias can be solved by the use of the Causal ATE metric.
CFL: Causally Fair Language Models Through Token-level Attribute Controlled Generation
Jun 01, 2023
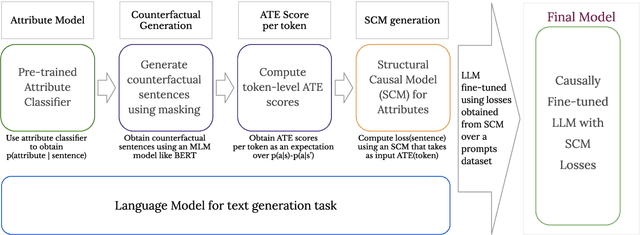
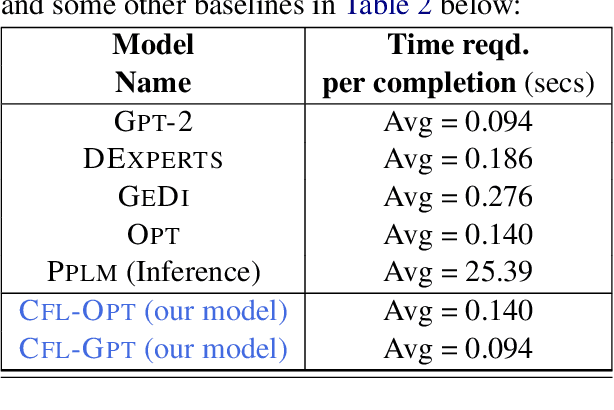
We propose a method to control the attributes of Language Models (LMs) for the text generation task using Causal Average Treatment Effect (ATE) scores and counterfactual augmentation. We explore this method, in the context of LM detoxification, and propose the Causally Fair Language (CFL) architecture for detoxifying pre-trained LMs in a plug-and-play manner. Our architecture is based on a Structural Causal Model (SCM) that is mathematically transparent and computationally efficient as compared with many existing detoxification techniques. We also propose several new metrics that aim to better understand the behaviour of LMs in the context of toxic text generation. Further, we achieve state of the art performance for toxic degeneration, which are computed using \RTP (RTP) benchmark. Our experiments show that CFL achieves such a detoxification without much impact on the model perplexity. We also show that CFL mitigates the unintended bias problem through experiments on the BOLD dataset.
Causal Graphs Underlying Generative Models: Path to Learning with Limited Data
Jul 14, 2022
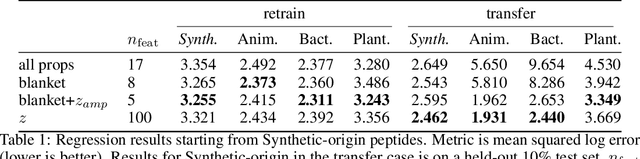
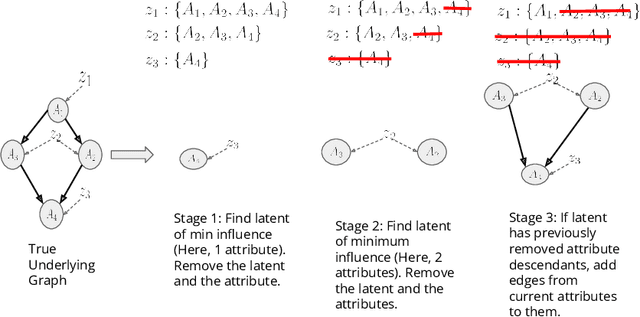
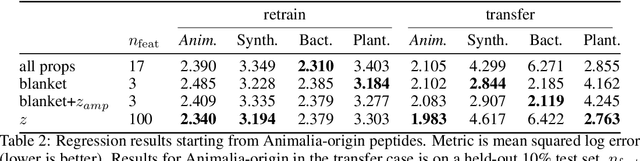
Training generative models that capture rich semantics of the data and interpreting the latent representations encoded by such models are very important problems in unsupervised learning. In this work, we provide a simple algorithm that relies on perturbation experiments on latent codes of a pre-trained generative autoencoder to uncover a causal graph that is implied by the generative model. We leverage pre-trained attribute classifiers and perform perturbation experiments to check for influence of a given latent variable on a subset of attributes. Given this, we show that one can fit an effective causal graph that models a structural equation model between latent codes taken as exogenous variables and attributes taken as observed variables. One interesting aspect is that a single latent variable controls multiple overlapping subsets of attributes unlike conventional approach that tries to impose full independence. Using a pre-trained RNN-based generative autoencoder trained on a dataset of peptide sequences, we demonstrate that the learnt causal graph from our algorithm between various attributes and latent codes can be used to predict a specific property for sequences which are unseen. We compare prediction models trained on either all available attributes or only the ones in the Markov blanket and empirically show that in both the unsupervised and supervised regimes, typically, using the predictor that relies on Markov blanket attributes generalizes better for out-of-distribution sequences.
Towards Interpreting Zoonotic Potential of Betacoronavirus Sequences With Attention
Aug 18, 2021
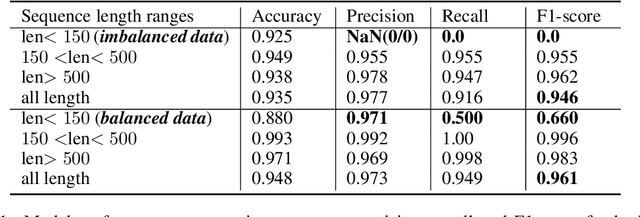
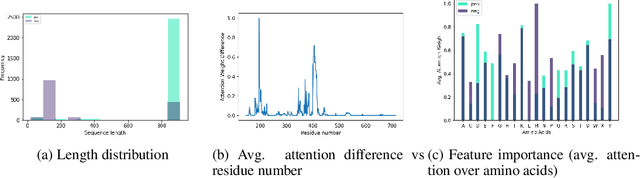
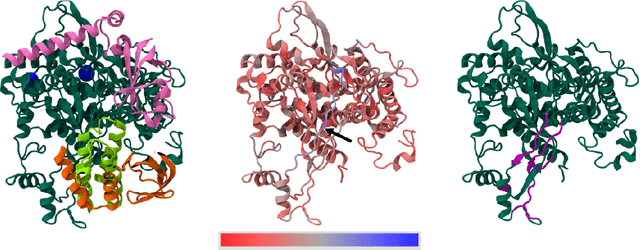
Current methods for viral discovery target evolutionarily conserved proteins that accurately identify virus families but remain unable to distinguish the zoonotic potential of newly discovered viruses. Here, we apply an attention-enhanced long-short-term memory (LSTM) deep neural net classifier to a highly conserved viral protein target to predict zoonotic potential across betacoronaviruses. The classifier performs with a 94% accuracy. Analysis and visualization of attention at the sequence and structure-level features indicate possible association between important protein-protein interactions governing viral replication in zoonotic betacoronaviruses and zoonotic transmission.
Optimizing Molecules using Efficient Queries from Property Evaluations
Nov 03, 2020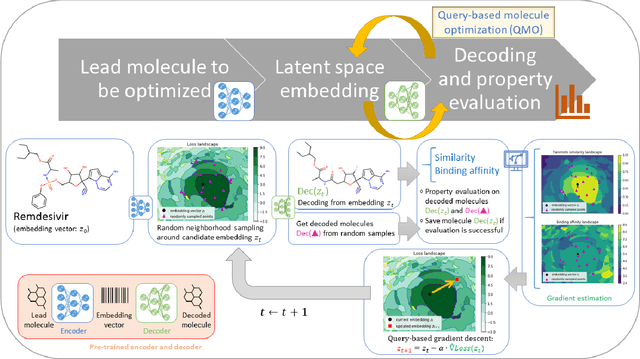
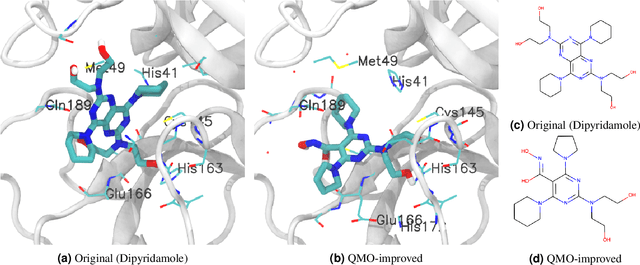
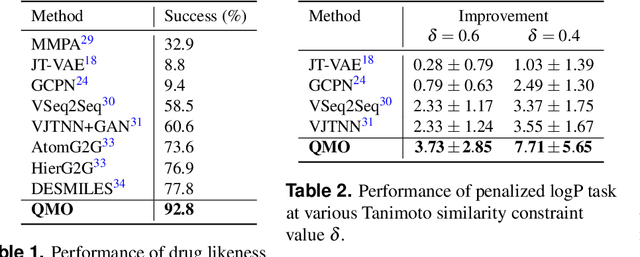
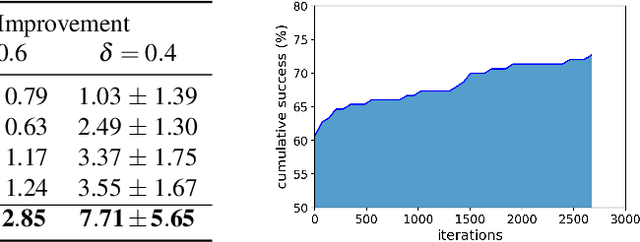
Machine learning has shown potential for optimizing existing molecules with more desirable properties, a critical step towards accelerating new chemical discovery. In this work, we propose QMO, a generic query-based molecule optimization framework that exploits latent embeddings from a molecule autoencoder. QMO improves the desired properties of an input molecule based on efficient queries, guided by a set of molecular property predictions and evaluation metrics. We show that QMO outperforms existing methods in the benchmark tasks of optimizing molecules for drug likeliness and solubility under similarity constraints. We also demonstrate significant property improvement using QMO on two new and challenging tasks that are also important in real-world discovery problems: (i) optimizing existing SARS-CoV-2 Main Protease inhibitors toward higher binding affinity; and (ii) improving known antimicrobial peptides towards lower toxicity. Results from QMO show high consistency with external validations, suggesting effective means of facilitating molecule optimization problems with design constraints.
Effects of Naturalistic Variation in Goal-Oriented Dialog
Oct 05, 2020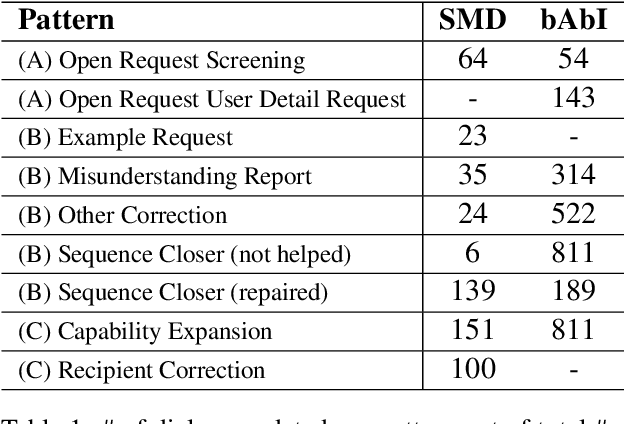
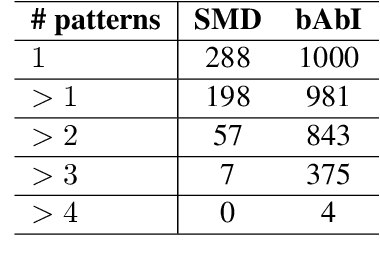
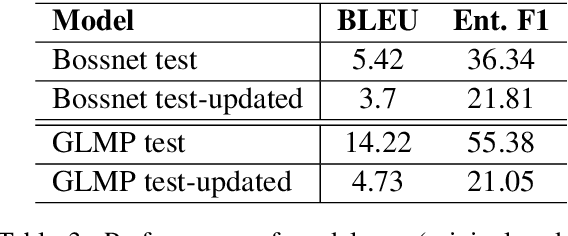
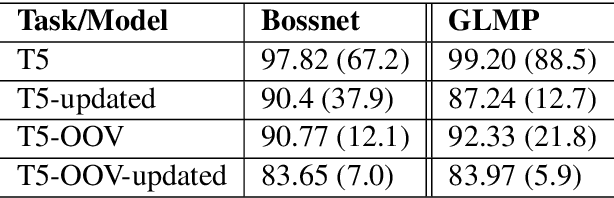
Existing benchmarks used to evaluate the performance of end-to-end neural dialog systems lack a key component: natural variation present in human conversations. Most datasets are constructed through crowdsourcing, where the crowd workers follow a fixed template of instructions while enacting the role of a user/agent. This results in straight-forward, somewhat routine, and mostly trouble-free conversations, as crowd workers do not think to represent the full range of actions that occur naturally with real users. In this work, we investigate the impact of naturalistic variation on two goal-oriented datasets: bAbI dialog task and Stanford Multi-Domain Dataset (SMD). We also propose new and more effective testbeds for both datasets, by introducing naturalistic variation by the user. We observe that there is a significant drop in performance (more than 60% in Ent. F1 on SMD and 85% in per-dialog accuracy on bAbI task) of recent state-of-the-art end-to-end neural methods such as BossNet and GLMP on both datasets.
Accelerating Antimicrobial Discovery with Controllable Deep Generative Models and Molecular Dynamics
May 22, 2020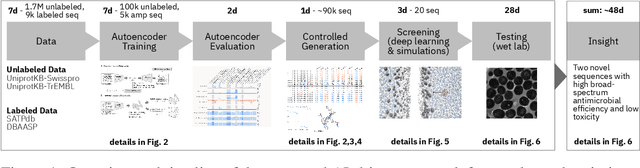
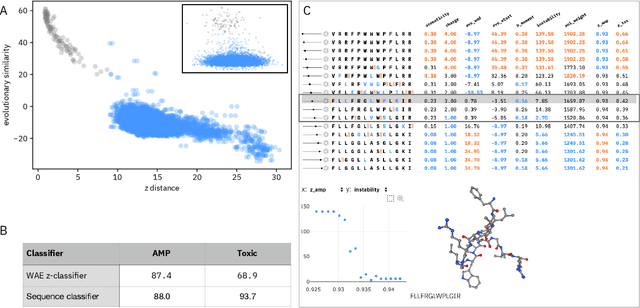
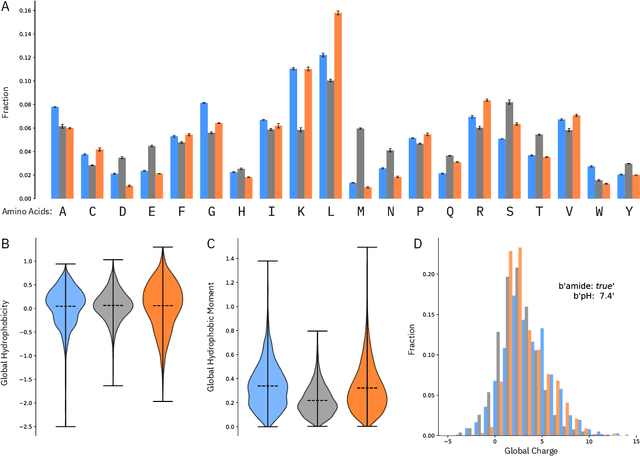
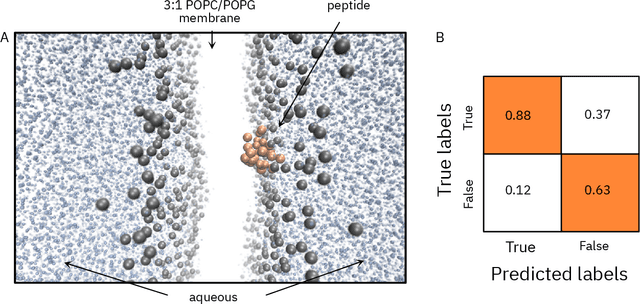
De novo therapeutic design is challenged by a vast chemical repertoire and multiple constraints such as high broad-spectrum potency and low toxicity. We propose CLaSS (Controlled Latent attribute Space Sampling) - a novel and efficient computational method for attribute-controlled generation of molecules, which leverages guidance from classifiers trained on an informative latent space of molecules modeled using a deep generative autoencoder. We further screen the generated molecules by using a set of deep learning classifiers in conjunction with novel physicochemical features derived from high-throughput molecular simulations. The proposed approach is employed for designing non-toxic antimicrobial peptides (AMPs) with strong broad-spectrum potency, which are emerging drug candidates for tackling antibiotic resistance. Synthesis and wet lab testing of only twenty designed sequences identified two novel and minimalist AMPs with high potency against diverse Gram-positive and Gram-negative pathogens, including the hard-to-treat multidrug-resistant K. pneumoniae, as well as low in vitro and in vivo toxicity. The proposed approach thus presents a viable path for faster discovery of potent and selective broad-spectrum antimicrobials with a higher success rate than state-of-the-art methods.
Co-regularized Alignment for Unsupervised Domain Adaptation
Nov 13, 2018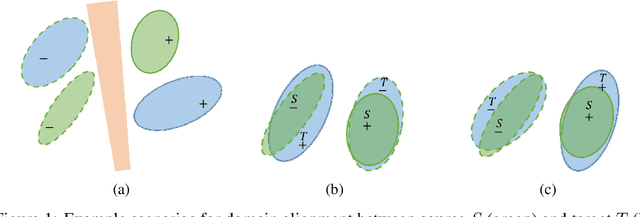
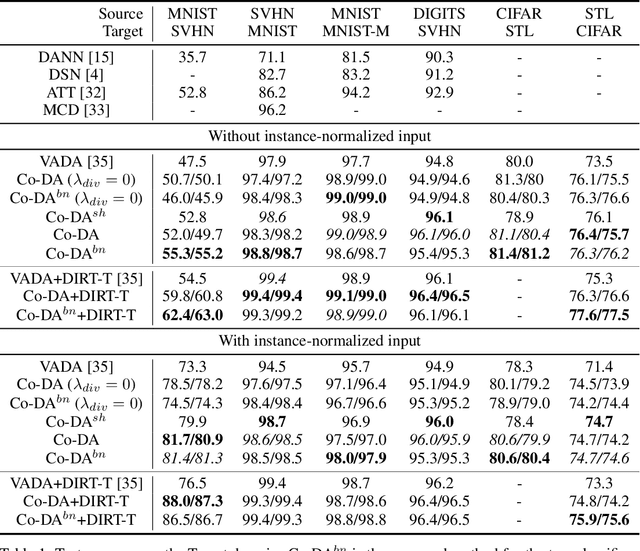
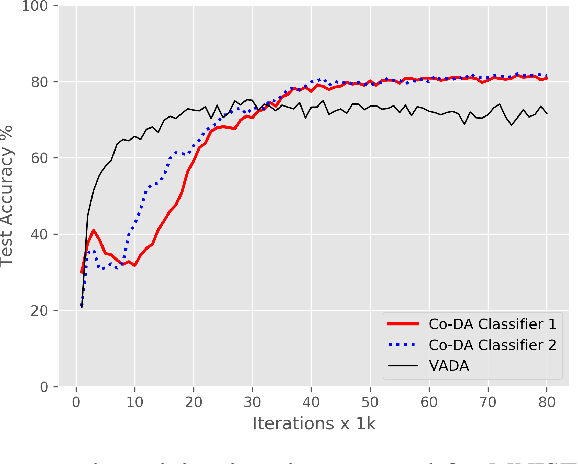
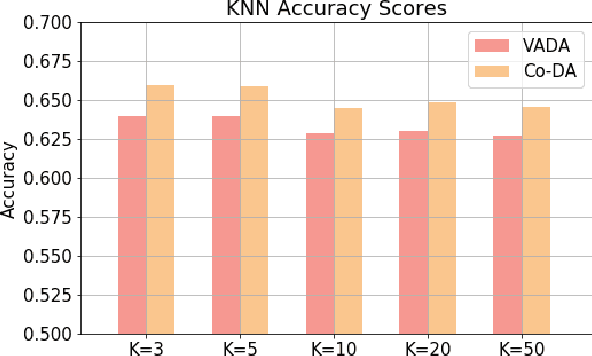
Deep neural networks, trained with large amount of labeled data, can fail to generalize well when tested with examples from a \emph{target domain} whose distribution differs from the training data distribution, referred as the \emph{source domain}. It can be expensive or even infeasible to obtain required amount of labeled data in all possible domains. Unsupervised domain adaptation sets out to address this problem, aiming to learn a good predictive model for the target domain using labeled examples from the source domain but only unlabeled examples from the target domain. Domain alignment approaches this problem by matching the source and target feature distributions, and has been used as a key component in many state-of-the-art domain adaptation methods. However, matching the marginal feature distributions does not guarantee that the corresponding class conditional distributions will be aligned across the two domains. We propose co-regularized domain alignment for unsupervised domain adaptation, which constructs multiple diverse feature spaces and aligns source and target distributions in each of them individually, while encouraging that alignments agree with each other with regard to the class predictions on the unlabeled target examples. The proposed method is generic and can be used to improve any domain adaptation method which uses domain alignment. We instantiate it in the context of a recent state-of-the-art method and observe that it provides significant performance improvements on several domain adaptation benchmarks.
PepCVAE: Semi-Supervised Targeted Design of Antimicrobial Peptide Sequences
Oct 22, 2018
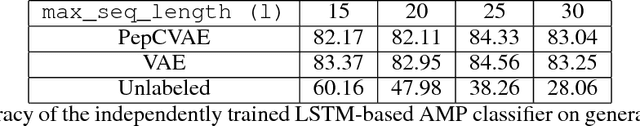
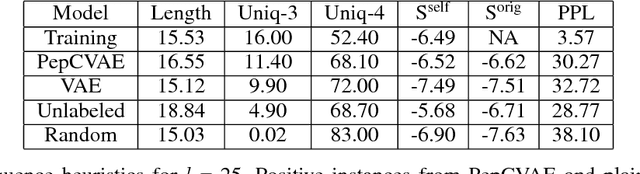
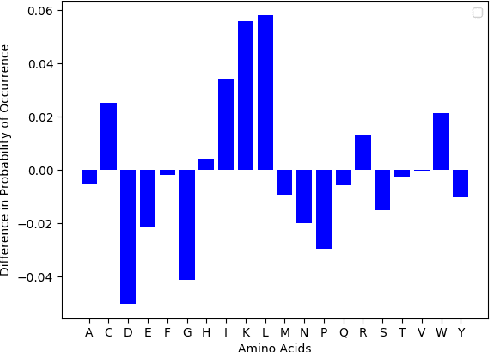
Given the emerging global threat of antimicrobial resistance, new methods for next-generation antimicrobial design are urgently needed. We report a peptide generation framework PepCVAE, based on a semi-supervised variational autoencoder (VAE) model, for designing novel antimicrobial peptide (AMP) sequences. Our model learns a rich latent space of the biological peptide context by taking advantage of abundant, unlabeled peptide sequences. The model further learns a disentangled antimicrobial attribute space by using the feedback from a jointly trained AMP classifier that uses limited labeled instances. The disentangled representation allows for controllable generation of AMPs. Extensive analysis of the PepCVAE-generated sequences reveals superior performance of our model in comparison to a plain VAE, as PepCVAE generates novel AMP sequences with higher long-range diversity, while being closer to the training distribution of biological peptides. These features are highly desired in next-generation antimicrobial design.
Improved Neural Text Attribute Transfer with Non-parallel Data
Dec 04, 2017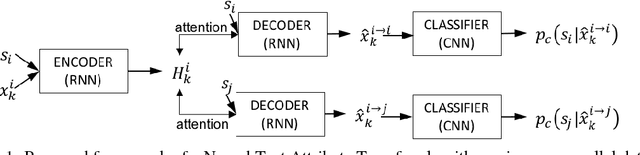
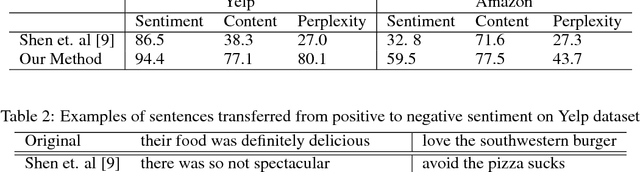
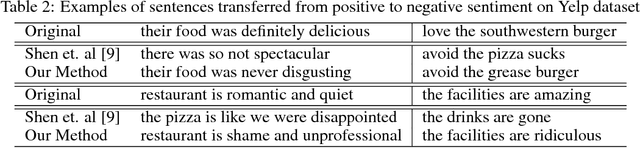
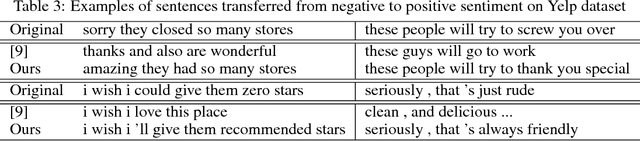
Text attribute transfer using non-parallel data requires methods that can perform disentanglement of content and linguistic attributes. In this work, we propose multiple improvements over the existing approaches that enable the encoder-decoder framework to cope with the text attribute transfer from non-parallel data. We perform experiments on the sentiment transfer task using two datasets. For both datasets, our proposed method outperforms a strong baseline in two of the three employed evaluation metrics.