 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAugmenting Molecular Deep Generative Models with Topological Data Analysis Representations
Jun 08, 2021


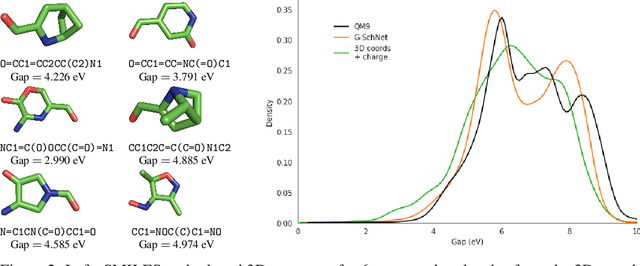
Deep generative models have emerged as a powerful tool for learning informative molecular representations and designing novel molecules with desired properties, with applications in drug discovery and material design. Deep generative auto-encoders defined over molecular SMILES strings have been a popular choice for that purpose. However, capturing salient molecular properties like quantum-chemical energies remains challenging and requires sophisticated neural net models of molecular graphs or geometry-based information. As a simpler and more efficient alternative, we present a SMILES Variational Auto-Encoder (VAE) augmented with topological data analysis (TDA) representations of molecules, known as persistence images. Our experiments show that this TDA augmentation enables a SMILES VAE to capture the complex relation between 3D geometry and electronic properties, and allows generation of novel, diverse, and valid molecules with geometric features consistent with the training data, which exhibit a varying range of global electronic structural properties, such as a small HOMO-LUMO gap - a critical property for designing organic solar cells. We demonstrate that our TDA augmentation yields better success in downstream tasks compared to models trained without these representations and can assist in targeted molecule discovery.
Optimizing Molecules using Efficient Queries from Property Evaluations
Nov 03, 2020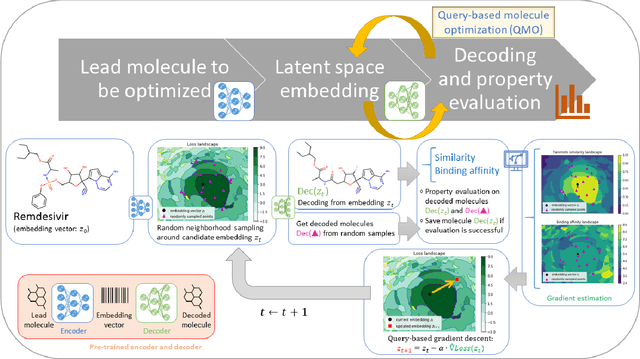
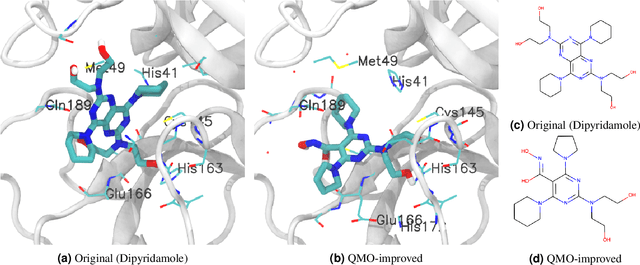
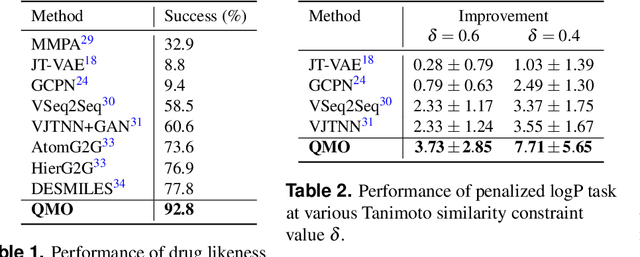
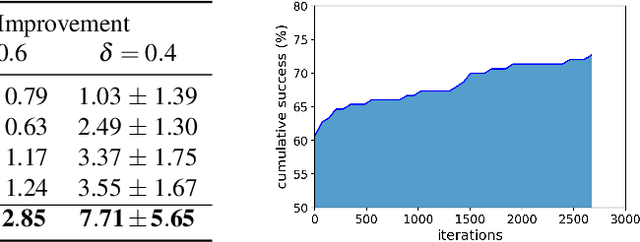
Machine learning has shown potential for optimizing existing molecules with more desirable properties, a critical step towards accelerating new chemical discovery. In this work, we propose QMO, a generic query-based molecule optimization framework that exploits latent embeddings from a molecule autoencoder. QMO improves the desired properties of an input molecule based on efficient queries, guided by a set of molecular property predictions and evaluation metrics. We show that QMO outperforms existing methods in the benchmark tasks of optimizing molecules for drug likeliness and solubility under similarity constraints. We also demonstrate significant property improvement using QMO on two new and challenging tasks that are also important in real-world discovery problems: (i) optimizing existing SARS-CoV-2 Main Protease inhibitors toward higher binding affinity; and (ii) improving known antimicrobial peptides towards lower toxicity. Results from QMO show high consistency with external validations, suggesting effective means of facilitating molecule optimization problems with design constraints.
Combinatorial Black-Box Optimization with Expert Advice
Jun 06, 2020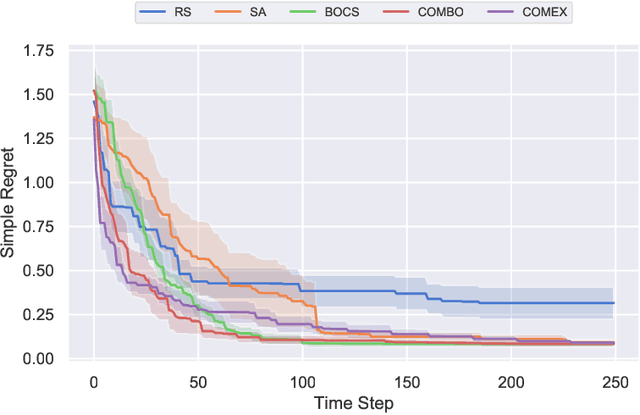
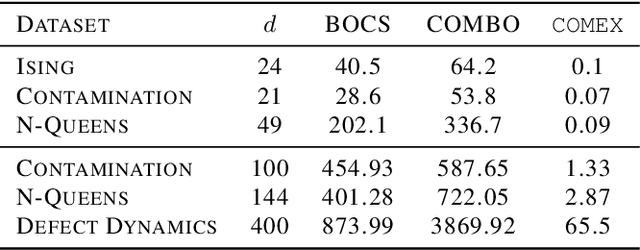
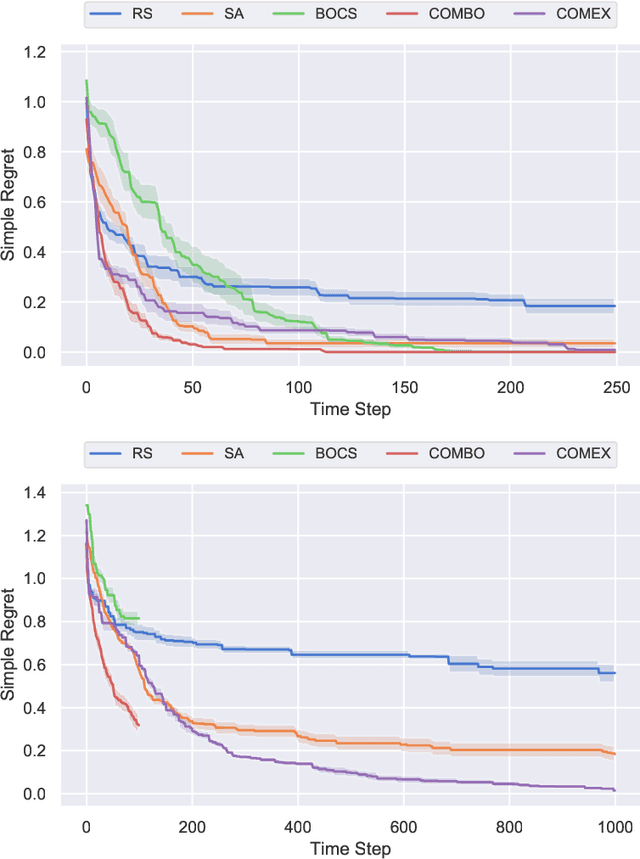
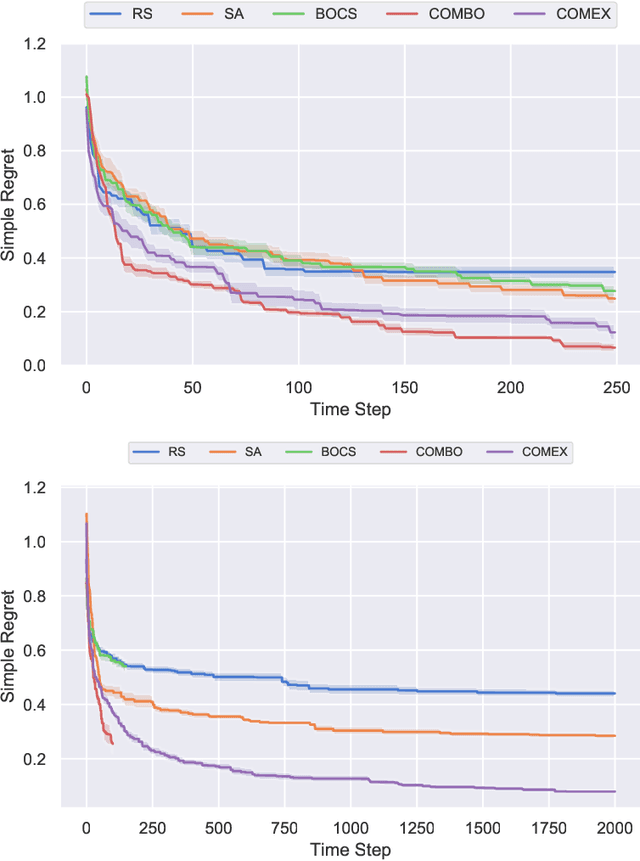
We consider the problem of black-box function optimization over the boolean hypercube. Despite the vast literature on black-box function optimization over continuous domains, not much attention has been paid to learning models for optimization over combinatorial domains until recently. However, the computational complexity of the recently devised algorithms are prohibitive even for moderate numbers of variables; drawing one sample using the existing algorithms is more expensive than a function evaluation for many black-box functions of interest. To address this problem, we propose a computationally efficient model learning algorithm based on multilinear polynomials and exponential weight updates. In the proposed algorithm, we alternate between simulated annealing with respect to the current polynomial representation and updating the weights using monomial experts' advice. Numerical experiments on various datasets in both unconstrained and sum-constrained boolean optimization indicate the competitive performance of the proposed algorithm, while improving the computational time up to several orders of magnitude compared to state-of-the-art algorithms in the literature.