 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking bandgap prediction in semiconductors under experimental and realistic evaluation settings
Apr 28, 2026Accurate bandgap prediction is crucial for semiconductor applications, yet machine learning models trained on computational data often struggle to generalize to experimental bandgap measurements. Challenges related to data fidelity, domain generalization, and model interpretability remain insufficiently addressed in existing evaluation frameworks. To bridge this gap, we introduce RealMat-BaG, a benchmark for assessing model reliability under experimentally relevant conditions. We curate an open-access dataset of experimental bandgaps with aligned crystal structures and compare graph neural networks as well as classical machine learning baselines. Our framework evaluates performance across statistical and domain-based splits, examines transfer from DFT-computed to experimental bandgaps, and analyzes interpretability at both elemental-property and structural levels. Our results reveal the fundamental generalization limitations of current bandgap prediction models and establish a benchmark aligned with experimental measurements for developing more reliable learning strategies for materials discovery.
Interpretable Multimodal Learning for Tumor Protein-Metal Binding: Progress, Challenges, and Perspectives
Apr 04, 2025In cancer therapeutics, protein-metal binding mechanisms critically govern drug pharmacokinetics and targeting efficacy, thereby fundamentally shaping the rational design of anticancer metallodrugs. While conventional laboratory methods used to study such mechanisms are often costly, low throughput, and limited in capturing dynamic biological processes, machine learning (ML) has emerged as a promising alternative. Despite increasing efforts to develop protein-metal binding datasets and ML algorithms, the application of ML in tumor protein-metal binding remains limited. Key challenges include a shortage of high-quality, tumor-specific datasets, insufficient consideration of multiple data modalities, and the complexity of interpreting results due to the ''black box'' nature of complex ML models. This paper summarizes recent progress and ongoing challenges in using ML to predict tumor protein-metal binding, focusing on data, modeling, and interpretability. We present multimodal protein-metal binding datasets and outline strategies for acquiring, curating, and preprocessing them for training ML models. Moreover, we explore the complementary value provided by different data modalities and examine methods for their integration. We also review approaches for improving model interpretability to support more trustworthy decisions in cancer research. Finally, we offer our perspective on research opportunities and propose strategies to address the scarcity of tumor protein data and the limited number of predictive models for tumor protein-metal binding. We also highlight two promising directions for effective metal-based drug design: integrating protein-protein interaction data to provide structural insights into metal-binding events and predicting structural changes in tumor proteins after metal binding.
Towards deployment-centric multimodal AI beyond vision and language
Apr 04, 2025Multimodal artificial intelligence (AI) integrates diverse types of data via machine learning to improve understanding, prediction, and decision-making across disciplines such as healthcare, science, and engineering. However, most multimodal AI advances focus on models for vision and language data, while their deployability remains a key challenge. We advocate a deployment-centric workflow that incorporates deployment constraints early to reduce the likelihood of undeployable solutions, complementing data-centric and model-centric approaches. We also emphasise deeper integration across multiple levels of multimodality and multidisciplinary collaboration to significantly broaden the research scope beyond vision and language. To facilitate this approach, we identify common multimodal-AI-specific challenges shared across disciplines and examine three real-world use cases: pandemic response, self-driving car design, and climate change adaptation, drawing expertise from healthcare, social science, engineering, science, sustainability, and finance. By fostering multidisciplinary dialogue and open research practices, our community can accelerate deployment-centric development for broad societal impact.
Co-evolution-based Metal-binding Residue Prediction with Graph Neural Networks
Feb 22, 2025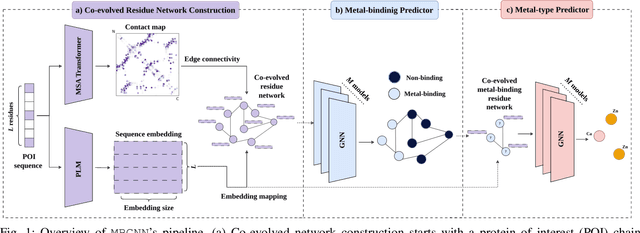
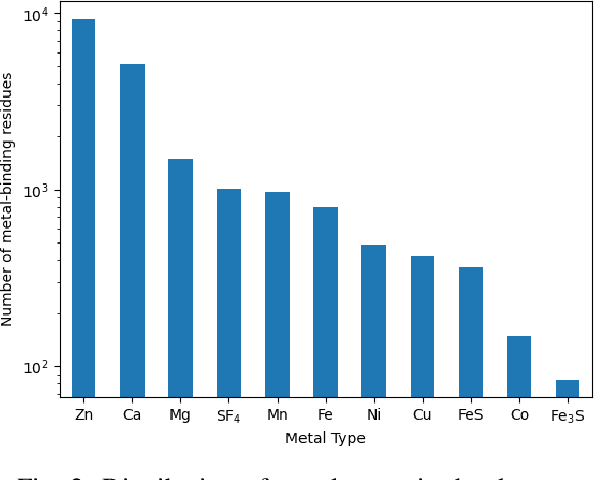
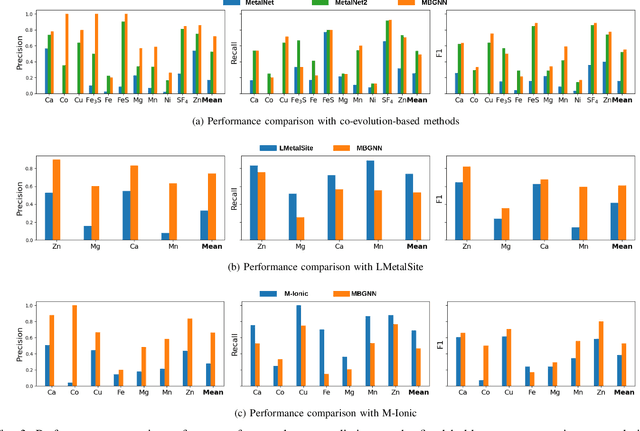
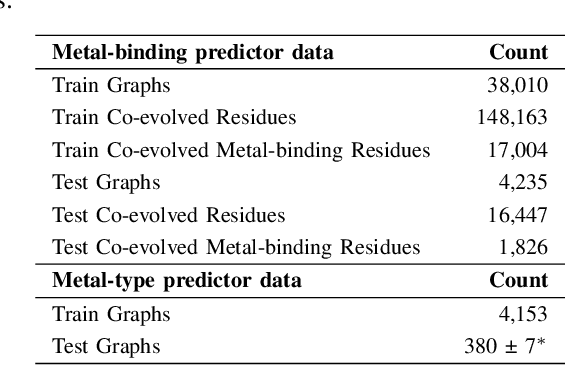
In computational structural biology, predicting metal-binding sites and their corresponding metal types is challenging due to the complexity of protein structures and interactions. Conventional sequence- and structure-based prediction approaches cannot capture the complex evolutionary relationships driving these interactions to facilitate understanding, while recent co-evolution-based approaches do not fully consider the entire structure of the co-evolved residue network. In this paper, we introduce MBGNN (Metal-Binding Graph Neural Network) that utilizes the entire co-evolved residue network and effectively captures the complex dependencies within protein structures via graph neural networks to enhance the prediction of co-evolved metal-binding residues and their associated metal types. Experimental results on a public dataset show that MBGNN outperforms existing co-evolution-based metal-binding prediction methods, and it is also competitive against recent sequence-based methods, showing the potential of integrating co-evolutionary insights with advanced machine learning to deepen our understanding of protein-metal interactions. The MBGNN code is publicly available at https://github.com/SRastegari/MBGNN.
Classifying the Stoichiometry of Virus-like Particles with Interpretable Machine Learning
Feb 17, 2025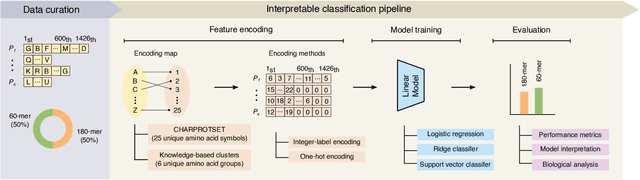

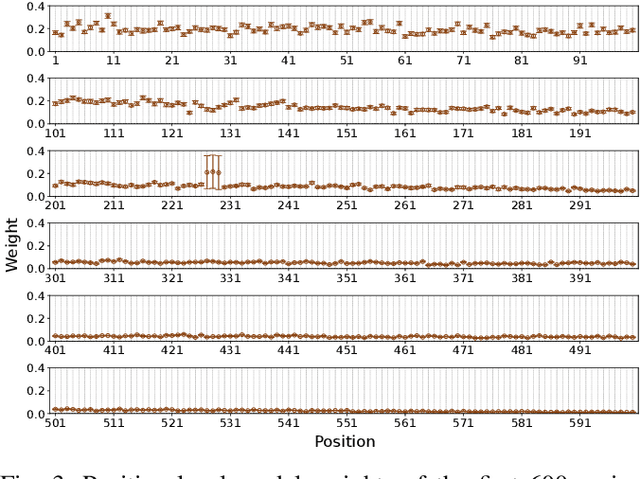
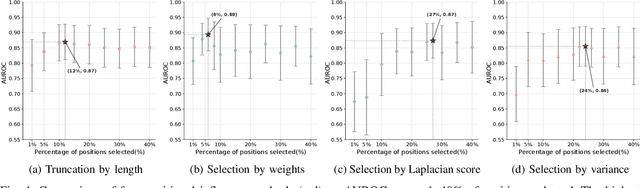
Virus-like particles (VLPs) are valuable for vaccine development due to their immune-triggering properties. Understanding their stoichiometry, the number of protein subunits to form a VLP, is critical for vaccine optimisation. However, current experimental methods to determine stoichiometry are time-consuming and require highly purified proteins. To efficiently classify stoichiometry classes in proteins, we curate a new dataset and propose an interpretable, data-driven pipeline leveraging linear machine learning models. We also explore the impact of feature encoding on model performance and interpretability, as well as methods to identify key protein sequence features influencing classification. The evaluation of our pipeline demonstrates that it can classify stoichiometry while revealing protein features that possibly influence VLP assembly. The data and code used in this work are publicly available at https://github.com/Shef-AIRE/StoicIML.
Mask prior-guided denoising diffusion improves inverse protein folding
Dec 10, 2024



Inverse protein folding generates valid amino acid sequences that can fold into a desired protein structure, with recent deep-learning advances showing significant potential and competitive performance. However, challenges remain in predicting highly uncertain regions, such as those with loops and disorders. To tackle such low-confidence residue prediction, we propose a \textbf{Ma}sk \textbf{p}rior-guided denoising \textbf{Diff}usion (\textbf{MapDiff}) framework that accurately captures both structural and residue interactions for inverse protein folding. MapDiff is a discrete diffusion probabilistic model that iteratively generates amino acid sequences with reduced noise, conditioned on a given protein backbone. To incorporate structural and residue interactions, we develop a graph-based denoising network with a mask prior pre-training strategy. Moreover, in the generative process, we combine the denoising diffusion implicit model with Monte-Carlo dropout to improve uncertainty estimation. Evaluation on four challenging sequence design benchmarks shows that MapDiff significantly outperforms state-of-the-art methods. Furthermore, the in-silico sequences generated by MapDiff closely resemble the physico-chemical and structural characteristics of native proteins across different protein families and architectures.
Investigating the Impact of Hard Samples on Accuracy Reveals In-class Data Imbalance
Sep 22, 2024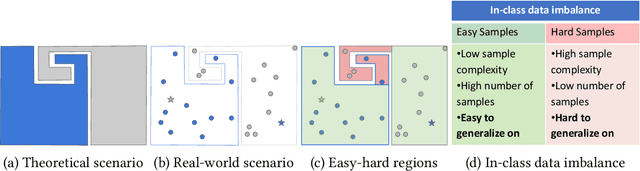
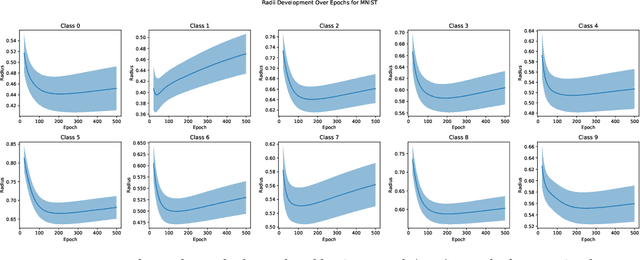
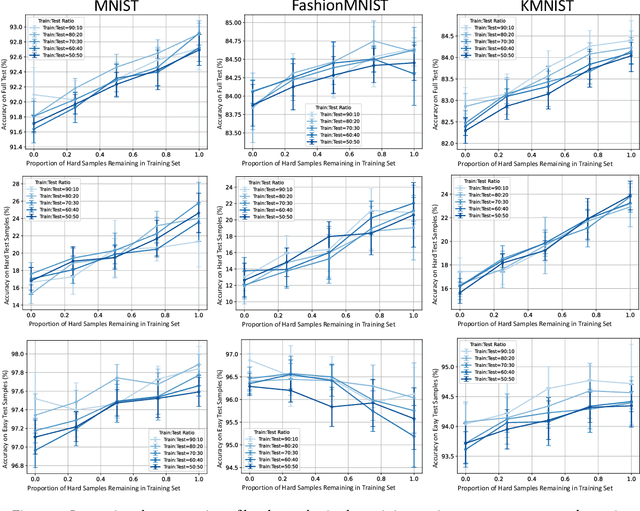
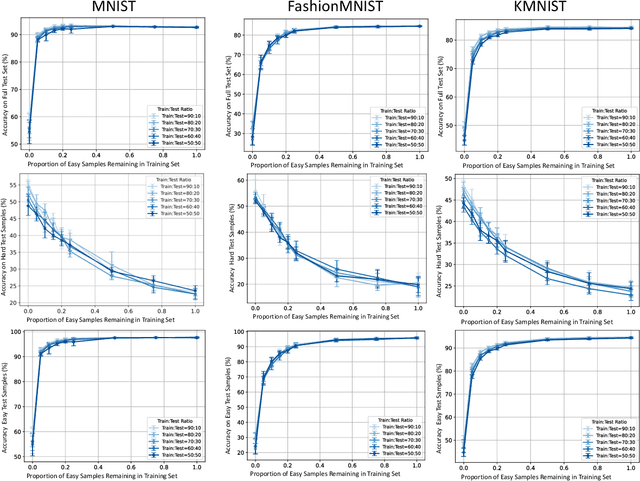
In the AutoML domain, test accuracy is heralded as the quintessential metric for evaluating model efficacy, underpinning a wide array of applications from neural architecture search to hyperparameter optimization. However, the reliability of test accuracy as the primary performance metric has been called into question, notably through research highlighting how label noise can obscure the true ranking of state-of-the-art models. We venture beyond, along another perspective where the existence of hard samples within datasets casts further doubt on the generalization capabilities inferred from test accuracy alone. Our investigation reveals that the distribution of hard samples between training and test sets affects the difficulty levels of those sets, thereby influencing the perceived generalization capability of models. We unveil two distinct generalization pathways-toward easy and hard samples-highlighting the complexity of achieving balanced model evaluation. Finally, we propose a benchmarking procedure for comparing hard sample identification methods, facilitating the advancement of more nuanced approaches in this area. Our primary goal is not to propose a definitive solution but to highlight the limitations of relying primarily on test accuracy as an evaluation metric, even when working with balanced datasets, by introducing the in-class data imbalance problem. By doing so, we aim to stimulate a critical discussion within the research community and open new avenues for research that consider a broader spectrum of model evaluation criteria. The anonymous code is available at https://github.com/PawPuk/CurvBIM blueunder the GPL-3.0 license.
Heterogeneous graph attention network improves cancer multiomics integration
Aug 05, 2024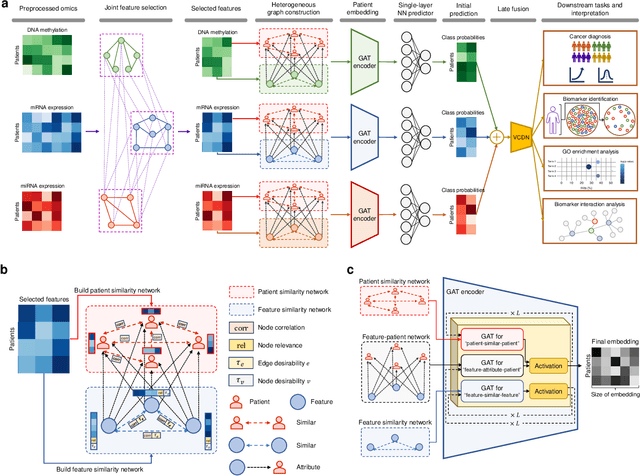
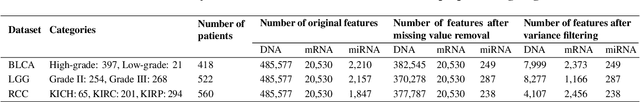
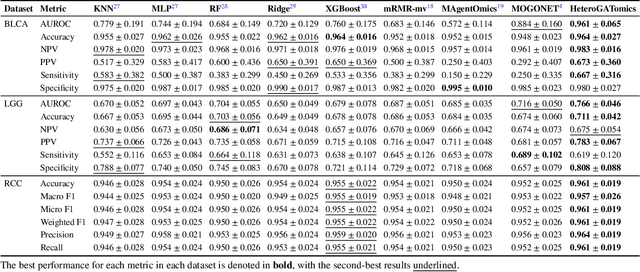
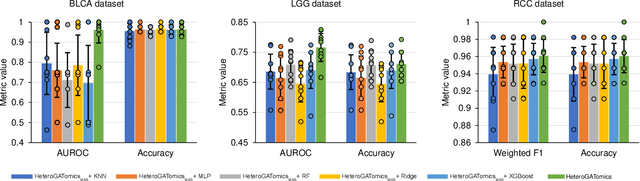
The increase in high-dimensional multiomics data demands advanced integration models to capture the complexity of human diseases. Graph-based deep learning integration models, despite their promise, struggle with small patient cohorts and high-dimensional features, often applying independent feature selection without modeling relationships among omics. Furthermore, conventional graph-based omics models focus on homogeneous graphs, lacking multiple types of nodes and edges to capture diverse structures. We introduce a Heterogeneous Graph ATtention network for omics integration (HeteroGATomics) to improve cancer diagnosis. HeteroGATomics performs joint feature selection through a multi-agent system, creating dedicated networks of feature and patient similarity for each omic modality. These networks are then combined into one heterogeneous graph for learning holistic omic-specific representations and integrating predictions across modalities. Experiments on three cancer multiomics datasets demonstrate HeteroGATomics' superior performance in cancer diagnosis. Moreover, HeteroGATomics enhances interpretability by identifying important biomarkers contributing to the diagnosis outcomes.
Group-specific discriminant analysis reveals statistically validated sex differences in lateralization of brain functional network
Apr 08, 2024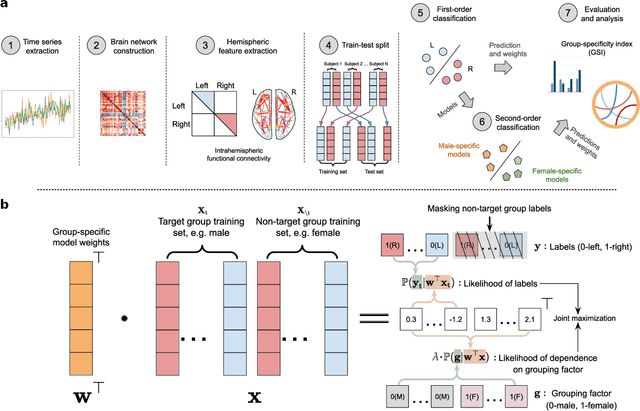

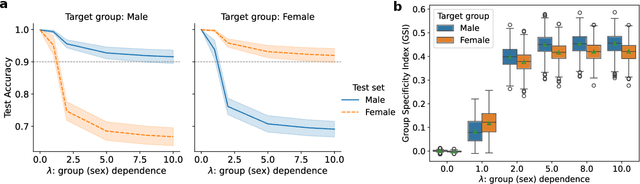
Lateralization is a fundamental feature of the human brain, where sex differences have been observed. Conventional studies in neuroscience on sex-specific lateralization are typically conducted on univariate statistical comparisons between male and female groups. However, these analyses often lack effective validation of group specificity. Here, we formulate modeling sex differences in lateralization of functional networks as a dual-classification problem, consisting of first-order classification for left vs. right functional networks and second-order classification for male vs. female models. To capture sex-specific patterns, we develop the Group-Specific Discriminant Analysis (GSDA) for first-order classification. The evaluation on two public neuroimaging datasets demonstrates the efficacy of GSDA in learning sex-specific models from functional networks, achieving a significant improvement in group specificity over baseline methods. The major sex differences are in the strength of lateralization and the interactions within and between lobes. The GSDA-based method is generic in nature and can be adapted to other group-specific analyses such as handedness-specific or disease-specific analyses.
Interpretable Multimodal Learning for Cardiovascular Hemodynamics Assessment
Apr 06, 2024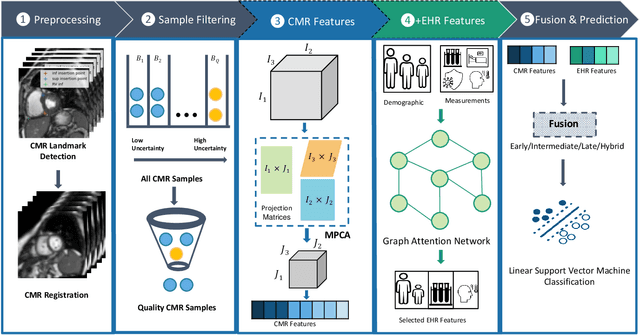
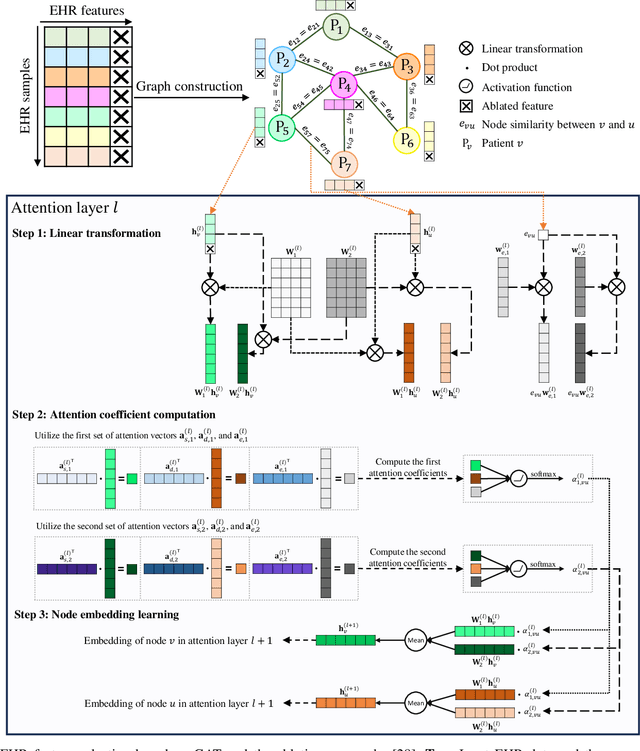
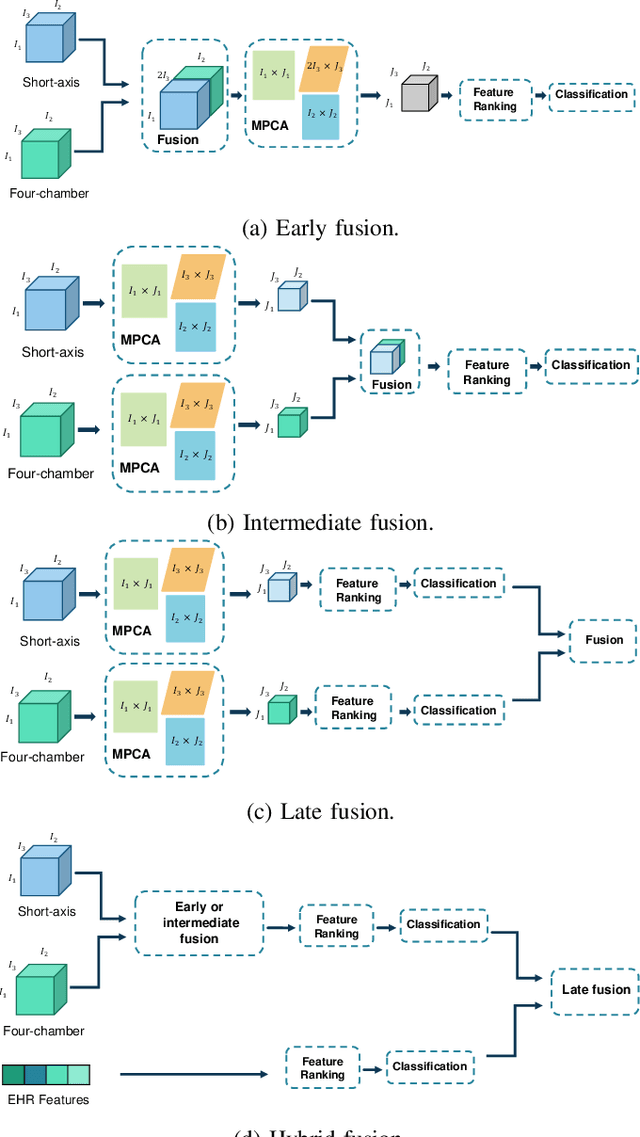
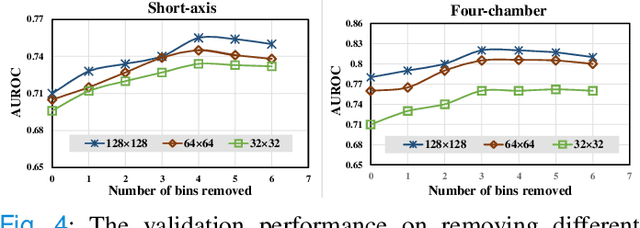
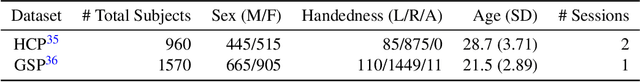
Pulmonary Arterial Wedge Pressure (PAWP) is an essential cardiovascular hemodynamics marker to detect heart failure. In clinical practice, Right Heart Catheterization is considered a gold standard for assessing cardiac hemodynamics while non-invasive methods are often needed to screen high-risk patients from a large population. In this paper, we propose a multimodal learning pipeline to predict PAWP marker. We utilize complementary information from Cardiac Magnetic Resonance Imaging (CMR) scans (short-axis and four-chamber) and Electronic Health Records (EHRs). We extract spatio-temporal features from CMR scans using tensor-based learning. We propose a graph attention network to select important EHR features for prediction, where we model subjects as graph nodes and feature relationships as graph edges using the attention mechanism. We design four feature fusion strategies: early, intermediate, late, and hybrid fusion. With a linear classifier and linear fusion strategies, our pipeline is interpretable. We validate our pipeline on a large dataset of $2,641$ subjects from our ASPIRE registry. The comparative study against state-of-the-art methods confirms the superiority of our pipeline. The decision curve analysis further validates that our pipeline can be applied to screen a large population. The code is available at https://github.com/prasunc/hemodynamics.