 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZeroFold: Protein-RNA Binding Affinity Predictions from Pre-Structural Embeddings
Mar 24, 2026The accurate prediction of protein-RNA binding affinity remains an unsolved problem in structural biology, limiting opportunities in understanding gene regulation and designing RNA-targeting therapeutics. A central obstacle is the structural flexibility of RNA, as, unlike proteins, RNA molecules exist as dynamic conformational ensembles. Thus, committing to a single predicted structure discards information relevant to binding. Here, we show that this obstacle can be addressed by extracting pre-structural embeddings, which are intermediate representations from a biomolecular foundation model captured before the structure decoding step. Pre-structural embeddings implicitly encode conformational ensemble information without requiring predicted structures. We build ZeroFold, a transformer-based model that combines pre-structural embeddings from Boltz-2 for both protein and RNA molecules through a cross-modal attention mechanism to predict binding affinity directly from sequence. To support training and evaluation, we construct PRADB, a curated dataset of 2,621 unique protein-RNA pairs with experimentally measured affinities drawn from four complementary databases. On a held-out test set constructed with 40% sequence identity thresholds, ZeroFold achieves a Spearman correlation of 0.65, a value approaching the ceiling imposed by experimental measurement noise. Under progressively fairer evaluation conditions that control for training-set overlap, ZeroFold compares favourably with respect to leading structure-based and leading sequence-based predictors, with the performance gap widening as sequence similarity to competitor training data is reduced. These results illustrate how pre-structural embeddings offer a representation strategy for flexible biomolecules, opening a route to affinity prediction for protein-RNA pairs for which no structural data exist.
Mask prior-guided denoising diffusion improves inverse protein folding
Dec 10, 2024



Inverse protein folding generates valid amino acid sequences that can fold into a desired protein structure, with recent deep-learning advances showing significant potential and competitive performance. However, challenges remain in predicting highly uncertain regions, such as those with loops and disorders. To tackle such low-confidence residue prediction, we propose a \textbf{Ma}sk \textbf{p}rior-guided denoising \textbf{Diff}usion (\textbf{MapDiff}) framework that accurately captures both structural and residue interactions for inverse protein folding. MapDiff is a discrete diffusion probabilistic model that iteratively generates amino acid sequences with reduced noise, conditioned on a given protein backbone. To incorporate structural and residue interactions, we develop a graph-based denoising network with a mask prior pre-training strategy. Moreover, in the generative process, we combine the denoising diffusion implicit model with Monte-Carlo dropout to improve uncertainty estimation. Evaluation on four challenging sequence design benchmarks shows that MapDiff significantly outperforms state-of-the-art methods. Furthermore, the in-silico sequences generated by MapDiff closely resemble the physico-chemical and structural characteristics of native proteins across different protein families and architectures.
PepINVENT: Generative peptide design beyond the natural amino acids
Sep 21, 2024
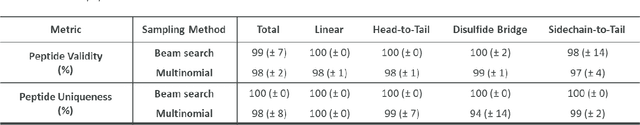

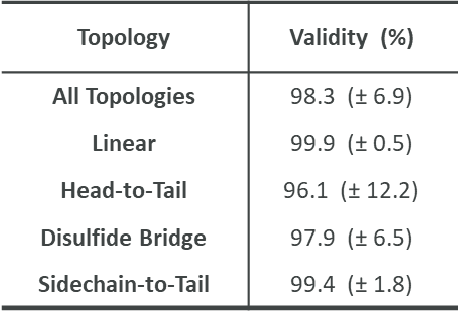
Peptides play a crucial role in the drug design and discovery whether as a therapeutic modality or a delivery agent. Non-natural amino acids (NNAAs) have been used to enhance the peptide properties from binding affinity, plasma stability to permeability. Incorporating novel NNAAs facilitates the design of more effective peptides with improved properties. The generative models used in the field, have focused on navigating the peptide sequence space. The sequence space is formed by combinations of a predefined set of amino acids. However, there is still a need for a tool to explore the peptide landscape beyond this enumerated space to unlock and effectively incorporate de novo design of new amino acids. To thoroughly explore the theoretical chemical space of the peptides, we present PepINVENT, a novel generative AI-based tool as an extension to the small molecule molecular design platform, REINVENT. PepINVENT navigates the vast space of natural and non-natural amino acids to propose valid, novel, and diverse peptide designs. The generative model can serve as a central tool for peptide-related tasks, as it was not trained on peptides with specific properties or topologies. The prior was trained to understand the granularity of peptides and to design amino acids for filling the masked positions within a peptide. PepINVENT coupled with reinforcement learning enables the goal-oriented design of peptides using its chemistry-informed generative capabilities. This study demonstrates PepINVENT's ability to explore the peptide space with unique and novel designs, and its capacity for property optimization in the context of therapeutically relevant peptides. Our tool can be employed for multi-parameter learning objectives, peptidomimetics, lead optimization, and variety of other tasks within the peptide domain.