 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBridging Chemists and AI: An Expert-Augmented Framework for Interpretable Route Evaluation
May 27, 2026Selecting efficient multi-step synthetic routes is a central challenge in organic synthesis, particularly in medicinal and process chemistry, where route choice directly impacts feasibility, cost, and development efficiency. Data-driven assessment systems often oversimplify the multi-objective nature of synthesis design and rely on proxy datasets, such as patent routes, rather than universally grounded criteria. To address this, we introduce an expert-augmented, data-driven scoring framework that integrates machine learning with chemists' domain knowledge for both numerical and explainable route assessment. A DeepSets-based model is trained using tree edit distance between reference and machine-generated routes, and then fine-tuned with expert evaluations to produce both quantitative scores and interpretable qualitative categories: Good, Plausible, and Bad. The resulting system achieves a Spearman correlation coefficient of 0.78 and a Pearson correlation of 0.77 for category assessment prediction, and 60.2% top-1 ranking accuracy for score prediction, substantially outperforming the previous baseline of 17.5%.
Confidence is the key: how conformal prediction enhances the generative design of permeable peptides
May 07, 2026Generative models coupled with reinforcement learning (RL), such as REINVENT and PepINVENT, have emerged as a powerful framework for de novo molecular design. During the ideation process these generative frameworks utilize various predictive models as part of the optimization objectives. However, the utility of the predictive models can be limited by their domain of applicability. When RL is used to explore the chemical space with predictive models, it can suggest molecules that lie outside the predictor's domain of applicability. As a result, the predictions may become less reliable, potentially steering designs into high reward but also high uncertainty chemical spaces. This is particularly pronounced for cyclic peptides which show therapeutic promise due to their modifiability and large interaction surfaces but are understudied compared to small molecules. While passive membrane permeation in cyclic peptides has attracted interest, identifying optimal permeable designs remains challenging yet crucial for targeting intracellular sites. We present an RL-guided generative framework that designs permeable cyclic peptides using an uncertainty-aware permeability predictor as the scoring component. To address predictive uncertainty, especially impacted by novel chemistry, we integrate conformal prediction (CP) as our uncertainty quantification method. CP assesses designs based on the calibrated model under a user-defined confidence level. We demonstrate that rewarding generated peptides with CP-informed predictions improves both reliability and efficiency of peptide optimization process. This also discourages exploration outside the predictor's applicability domain. This approach bridges the gap between predictive uncertainty and RL-guided exploration, showing how generative modelling and conformal prediction can be combined for the first time.
Temporal Distribution Shift in Real-World Pharmaceutical Data: Implications for Uncertainty Quantification in QSAR Models
Feb 06, 2025



The estimation of uncertainties associated with predictions from quantitative structure-activity relationship (QSAR) models can accelerate the drug discovery process by identifying promising experiments and allowing an efficient allocation of resources. Several computational tools exist that estimate the predictive uncertainty in machine learning models. However, deviations from the i.i.d. setting have been shown to impair the performance of these uncertainty quantification methods. We use a real-world pharmaceutical dataset to address the pressing need for a comprehensive, large-scale evaluation of uncertainty estimation methods in the context of realistic distribution shifts over time. We investigate the performance of several uncertainty estimation methods, including ensemble-based and Bayesian approaches. Furthermore, we use this real-world setting to systematically assess the distribution shifts in label and descriptor space and their impact on the capability of the uncertainty estimation methods. Our study reveals significant shifts over time in both label and descriptor space and a clear connection between the magnitude of the shift and the nature of the assay. Moreover, we show that pronounced distribution shifts impair the performance of popular uncertainty estimation methods used in QSAR models. This work highlights the challenges of identifying uncertainty quantification methods that remain reliable under distribution shifts introduced by real-world data.
Publishing Neural Networks in Drug Discovery Might Compromise Training Data Privacy
Oct 22, 2024



This study investigates the risks of exposing confidential chemical structures when machine learning models trained on these structures are made publicly available. We use membership inference attacks, a common method to assess privacy that is largely unexplored in the context of drug discovery, to examine neural networks for molecular property prediction in a black-box setting. Our results reveal significant privacy risks across all evaluated datasets and neural network architectures. Combining multiple attacks increases these risks. Molecules from minority classes, often the most valuable in drug discovery, are particularly vulnerable. We also found that representing molecules as graphs and using message-passing neural networks may mitigate these risks. We provide a framework to assess privacy risks of classification models and molecular representations. Our findings highlight the need for careful consideration when sharing neural networks trained on proprietary chemical structures, informing organisations and researchers about the trade-offs between data confidentiality and model openness.
Diversity-Aware Reinforcement Learning for de novo Drug Design
Oct 14, 2024



Fine-tuning a pre-trained generative model has demonstrated good performance in generating promising drug molecules. The fine-tuning task is often formulated as a reinforcement learning problem, where previous methods efficiently learn to optimize a reward function to generate potential drug molecules. Nevertheless, in the absence of an adaptive update mechanism for the reward function, the optimization process can become stuck in local optima. The efficacy of the optimal molecule in a local optimization may not translate to usefulness in the subsequent drug optimization process or as a potential standalone clinical candidate. Therefore, it is important to generate a diverse set of promising molecules. Prior work has modified the reward function by penalizing structurally similar molecules, primarily focusing on finding molecules with higher rewards. To date, no study has comprehensively examined how different adaptive update mechanisms for the reward function influence the diversity of generated molecules. In this work, we investigate a wide range of intrinsic motivation methods and strategies to penalize the extrinsic reward, and how they affect the diversity of the set of generated molecules. Our experiments reveal that combining structure- and prediction-based methods generally yields better results in terms of molecular diversity.
PepINVENT: Generative peptide design beyond the natural amino acids
Sep 21, 2024
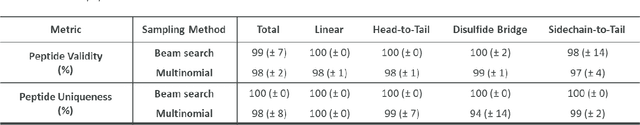

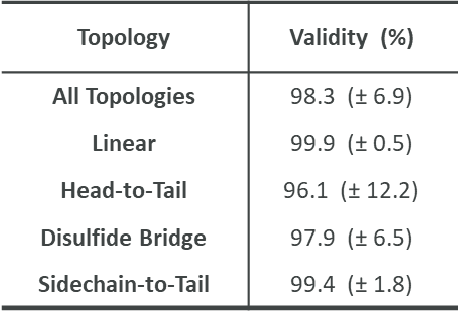
Peptides play a crucial role in the drug design and discovery whether as a therapeutic modality or a delivery agent. Non-natural amino acids (NNAAs) have been used to enhance the peptide properties from binding affinity, plasma stability to permeability. Incorporating novel NNAAs facilitates the design of more effective peptides with improved properties. The generative models used in the field, have focused on navigating the peptide sequence space. The sequence space is formed by combinations of a predefined set of amino acids. However, there is still a need for a tool to explore the peptide landscape beyond this enumerated space to unlock and effectively incorporate de novo design of new amino acids. To thoroughly explore the theoretical chemical space of the peptides, we present PepINVENT, a novel generative AI-based tool as an extension to the small molecule molecular design platform, REINVENT. PepINVENT navigates the vast space of natural and non-natural amino acids to propose valid, novel, and diverse peptide designs. The generative model can serve as a central tool for peptide-related tasks, as it was not trained on peptides with specific properties or topologies. The prior was trained to understand the granularity of peptides and to design amino acids for filling the masked positions within a peptide. PepINVENT coupled with reinforcement learning enables the goal-oriented design of peptides using its chemistry-informed generative capabilities. This study demonstrates PepINVENT's ability to explore the peptide space with unique and novel designs, and its capacity for property optimization in the context of therapeutically relevant peptides. Our tool can be employed for multi-parameter learning objectives, peptidomimetics, lead optimization, and variety of other tasks within the peptide domain.
Enhancing Uncertainty Quantification in Drug Discovery with Censored Regression Labels
Sep 06, 2024



In the early stages of drug discovery, decisions regarding which experiments to pursue can be influenced by computational models. These decisions are critical due to the time-consuming and expensive nature of the experiments. Therefore, it is becoming essential to accurately quantify the uncertainty in machine learning predictions, such that resources can be used optimally and trust in the models improves. While computational methods for drug discovery often suffer from limited data and sparse experimental observations, additional information can exist in the form of censored labels that provide thresholds rather than precise values of observations. However, the standard approaches that quantify uncertainty in machine learning cannot fully utilize censored labels. In this work, we adapt ensemble-based, Bayesian, and Gaussian models with tools to learn from censored labels by using the Tobit model from survival analysis. Our results demonstrate that despite the partial information available in censored labels, they are essential to accurately and reliably model the real pharmaceutical setting.
Achieving Well-Informed Decision-Making in Drug Discovery: A Comprehensive Calibration Study using Neural Network-Based Structure-Activity Models
Jul 19, 2024



In the drug discovery process, where experiments can be costly and time-consuming, computational models that predict drug-target interactions are valuable tools to accelerate the development of new therapeutic agents. Estimating the uncertainty inherent in these neural network predictions provides valuable information that facilitates optimal decision-making when risk assessment is crucial. However, such models can be poorly calibrated, which results in unreliable uncertainty estimates that do not reflect the true predictive uncertainty. In this study, we compare different metrics, including accuracy and calibration scores, used for model hyperparameter tuning to investigate which model selection strategy achieves well-calibrated models. Furthermore, we propose to use a computationally efficient Bayesian uncertainty estimation method named Bayesian Linear Probing (BLP), which generates Hamiltonian Monte Carlo (HMC) trajectories to obtain samples for the parameters of a Bayesian Logistic Regression fitted to the hidden layer of the baseline neural network. We report that BLP improves model calibration and achieves the performance of common uncertainty quantification methods by combining the benefits of uncertainty estimation and probability calibration methods. Finally, we show that combining post hoc calibration method with well-performing uncertainty quantification approaches can boost model accuracy and calibration.
Utilizing Reinforcement Learning for de novo Drug Design
Mar 30, 2023Deep learning-based approaches for generating novel drug molecules with specific properties have gained a lot of interest in the last years. Recent studies have demonstrated promising performance for string-based generation of novel molecules utilizing reinforcement learning. In this paper, we develop a unified framework for using reinforcement learning for de novo drug design, wherein we systematically study various on- and off-policy reinforcement learning algorithms and replay buffers to learn an RNN-based policy to generate novel molecules predicted to be active against the dopamine receptor DRD2. Our findings suggest that it is advantageous to use at least both top-scoring and low-scoring molecules for updating the policy when structural diversity is essential. Using all generated molecules at an iteration seems to enhance performance stability for on-policy algorithms. In addition, when replaying high, intermediate, and low-scoring molecules, off-policy algorithms display the potential of improving the structural diversity and number of active molecules generated, but possibly at the cost of a longer exploration phase. Our work provides an open-source framework enabling researchers to investigate various reinforcement learning methods for de novo drug design.
Industry-Scale Orchestrated Federated Learning for Drug Discovery
Oct 17, 2022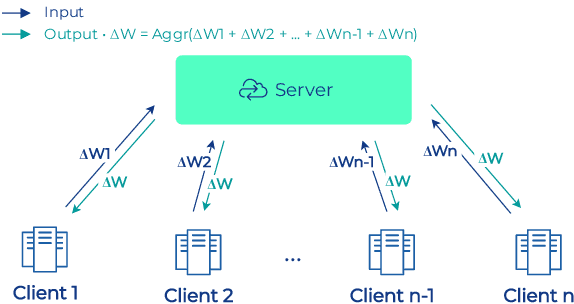
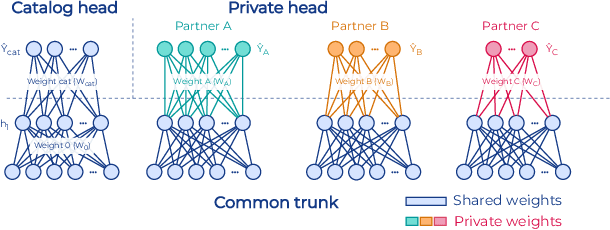
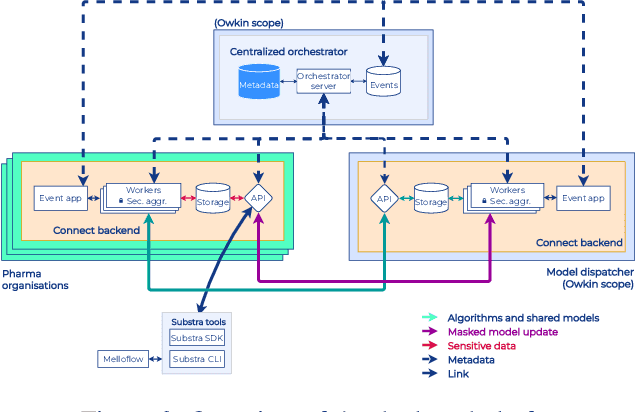
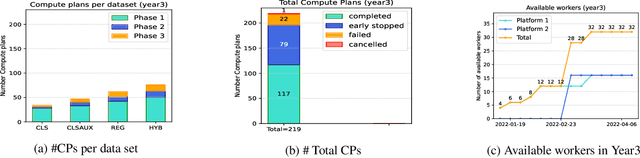
To apply federated learning to drug discovery we developed a novel platform in the context of European Innovative Medicines Initiative (IMI) project MELLODDY (grant n{\deg}831472), which was comprised of 10 pharmaceutical companies, academic research labs, large industrial companies and startups. To the best of our knowledge, The MELLODDY platform was the first industry-scale platform to enable the creation of a global federated model for drug discovery without sharing the confidential data sets of the individual partners. The federated model was trained on the platform by aggregating the gradients of all contributing partners in a cryptographic, secure way following each training iteration. The platform was deployed on an Amazon Web Services (AWS) multi-account architecture running Kubernetes clusters in private subnets. Organisationally, the roles of the different partners were codified as different rights and permissions on the platform and administrated in a decentralized way. The MELLODDY platform generated new scientific discoveries which are described in a companion paper.