 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFederated Block-Term Tensor Regression for decentralised data analysis in healthcare
Dec 02, 2024Block-Term Tensor Regression (BTTR) has proven to be a powerful tool for modeling complex, high-dimensional data by leveraging multilinear relationships, making it particularly well-suited for applications in healthcare and neuroscience. However, traditional implementations of BTTR rely on centralized datasets, which pose significant privacy risks and hinder collaboration across institutions. To address these challenges, we introduce Federated Block-Term Tensor Regression (FBTTR), an extension of BTTR designed for federated learning scenarios. FBTTR enables decentralized data analysis, allowing institutions to collaboratively build predictive models while preserving data privacy and complying with regulations. FBTTR represents a major step forward in applying tensor regression to federated learning environments. Its performance is evaluated in two case studies: finger movement decoding from Electrocorticography (ECoG) signals and heart disease prediction. In the first case study, using the BCI Competition IV dataset, FBTTR outperforms non-multilinear models, demonstrating superior accuracy in decoding finger movements. For the dataset, for subject 3, the thumb obtained a performance of 0.76 $\pm$ .05 compared to 0.71 $\pm$ 0.05 for centralised BTTR. In the second case study, FBTTR is applied to predict heart disease using real-world clinical datasets, outperforming both standard federated learning approaches and centralized BTTR models. In the Fed-Heart-Disease Dataset, an AUC-ROC was obtained of 0.872 $\pm$ 0.02 and an accuracy of 0.772 $\pm$ 0.02 compared to 0.812 $\pm$ 0.003 and 0.753 $\pm$ 0.007 for the centralized model.
Achieving Well-Informed Decision-Making in Drug Discovery: A Comprehensive Calibration Study using Neural Network-Based Structure-Activity Models
Jul 19, 2024



In the drug discovery process, where experiments can be costly and time-consuming, computational models that predict drug-target interactions are valuable tools to accelerate the development of new therapeutic agents. Estimating the uncertainty inherent in these neural network predictions provides valuable information that facilitates optimal decision-making when risk assessment is crucial. However, such models can be poorly calibrated, which results in unreliable uncertainty estimates that do not reflect the true predictive uncertainty. In this study, we compare different metrics, including accuracy and calibration scores, used for model hyperparameter tuning to investigate which model selection strategy achieves well-calibrated models. Furthermore, we propose to use a computationally efficient Bayesian uncertainty estimation method named Bayesian Linear Probing (BLP), which generates Hamiltonian Monte Carlo (HMC) trajectories to obtain samples for the parameters of a Bayesian Logistic Regression fitted to the hidden layer of the baseline neural network. We report that BLP improves model calibration and achieves the performance of common uncertainty quantification methods by combining the benefits of uncertainty estimation and probability calibration methods. Finally, we show that combining post hoc calibration method with well-performing uncertainty quantification approaches can boost model accuracy and calibration.
Atom-Level Optical Chemical Structure Recognition with Limited Supervision
Apr 02, 2024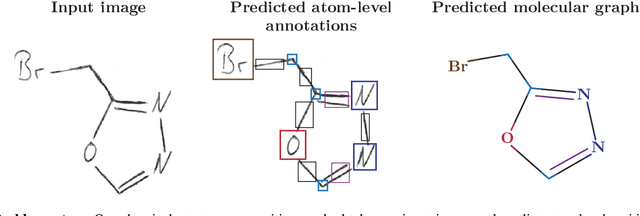


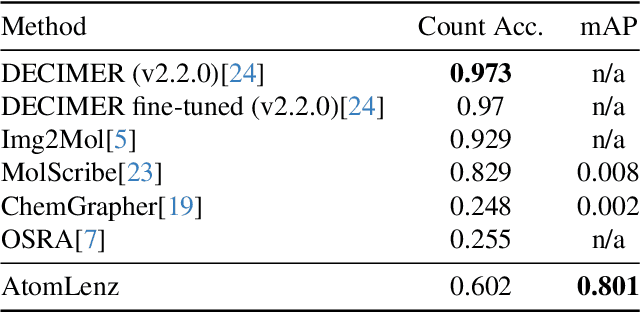
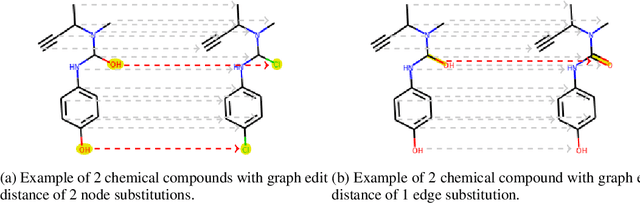
Identifying the chemical structure from a graphical representation, or image, of a molecule is a challenging pattern recognition task that would greatly benefit drug development. Yet, existing methods for chemical structure recognition do not typically generalize well, and show diminished effectiveness when confronted with domains where data is sparse, or costly to generate, such as hand-drawn molecule images. To address this limitation, we propose a new chemical structure recognition tool that delivers state-of-the-art performance and can adapt to new domains with a limited number of data samples and supervision. Unlike previous approaches, our method provides atom-level localization, and can therefore segment the image into the different atoms and bonds. Our model is the first model to perform OCSR with atom-level entity detection with only SMILES supervision. Through rigorous and extensive benchmarking, we demonstrate the preeminence of our chemical structure recognition approach in terms of data efficiency, accuracy, and atom-level entity prediction.
How good Neural Networks interpretation methods really are? A quantitative benchmark
Apr 05, 2023



Saliency Maps (SMs) have been extensively used to interpret deep learning models decision by highlighting the features deemed relevant by the model. They are used on highly nonlinear problems, where linear feature selection (FS) methods fail at highlighting relevant explanatory variables. However, the reliability of gradient-based feature attribution methods such as SM has mostly been only qualitatively (visually) assessed, and quantitative benchmarks are currently missing, partially due to the lack of a definite ground truth on image data. Concerned about the apophenic biases introduced by visual assessment of these methods, in this paper we propose a synthetic quantitative benchmark for Neural Networks (NNs) interpretation methods. For this purpose, we built synthetic datasets with nonlinearly separable classes and increasing number of decoy (random) features, illustrating the challenge of FS in high-dimensional settings. We also compare these methods to conventional approaches such as mRMR or Random Forests. Our results show that our simple synthetic datasets are sufficient to challenge most of the benchmarked methods. TreeShap, mRMR and LassoNet are the best performing FS methods. We also show that, when quantifying the relevance of a few non linearly-entangled predictive features diluted in a large number of irrelevant noisy variables, neural network-based FS and interpretation methods are still far from being reliable.
Weakly Supervised Knowledge Transfer with Probabilistic Logical Reasoning for Object Detection
Mar 09, 2023



Training object detection models usually requires instance-level annotations, such as the positions and labels of all objects present in each image. Such supervision is unfortunately not always available and, more often, only image-level information is provided, also known as weak supervision. Recent works have addressed this limitation by leveraging knowledge from a richly annotated domain. However, the scope of weak supervision supported by these approaches has been very restrictive, preventing them to use all available information. In this work, we propose ProbKT, a framework based on probabilistic logical reasoning that allows to train object detection models with arbitrary types of weak supervision. We empirically show on different datasets that using all available information is beneficial as our ProbKT leads to significant improvement on target domain and better generalization compared to existing baselines. We also showcase the ability of our approach to handle complex logic statements as supervision signal.
Industry-Scale Orchestrated Federated Learning for Drug Discovery
Oct 17, 2022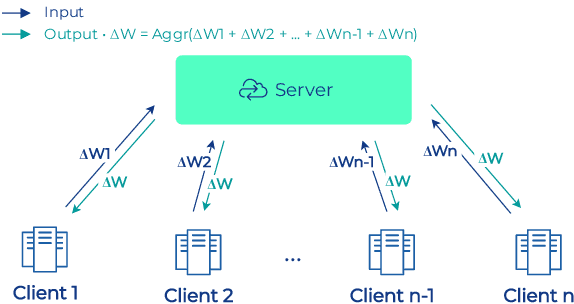
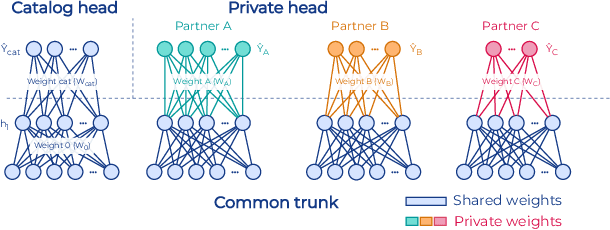
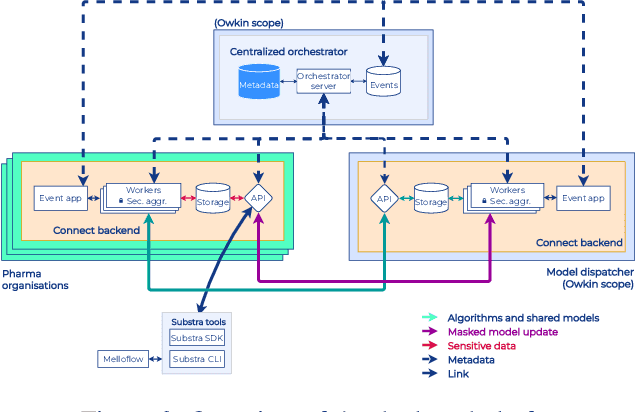
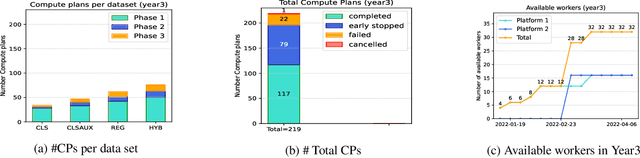
To apply federated learning to drug discovery we developed a novel platform in the context of European Innovative Medicines Initiative (IMI) project MELLODDY (grant n{\deg}831472), which was comprised of 10 pharmaceutical companies, academic research labs, large industrial companies and startups. To the best of our knowledge, The MELLODDY platform was the first industry-scale platform to enable the creation of a global federated model for drug discovery without sharing the confidential data sets of the individual partners. The federated model was trained on the platform by aggregating the gradients of all contributing partners in a cryptographic, secure way following each training iteration. The platform was deployed on an Amazon Web Services (AWS) multi-account architecture running Kubernetes clusters in private subnets. Organisationally, the roles of the different partners were codified as different rights and permissions on the platform and administrated in a decentralized way. The MELLODDY platform generated new scientific discoveries which are described in a companion paper.
SparseChem: Fast and accurate machine learning model for small molecules
Mar 09, 2022SparseChem provides fast and accurate machine learning models for biochemical applications. Especially, the package supports very high-dimensional sparse inputs, e.g., millions of features and millions of compounds. It is possible to train classification, regression and censored regression models, or combination of them from command line. Additionally, the library can be accessed directly from Python. Source code and documentation is freely available under MIT License on GitHub.
Self-Labeling of Fully Mediating Representations by Graph Alignment
Mar 25, 2021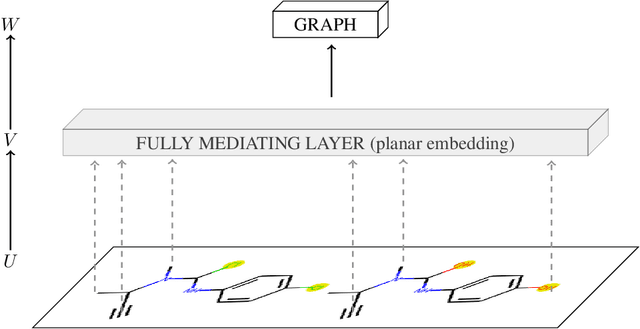

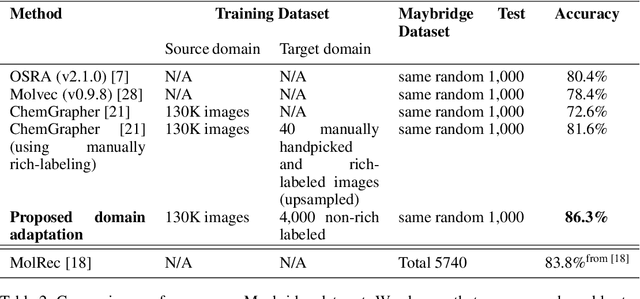
To be able to predict a molecular graph structure ($W$) given a 2D image of a chemical compound ($U$) is a challenging problem in machine learning. We are interested to learn $f: U \rightarrow W$ where we have a fully mediating representation $V$ such that $f$ factors into $U \rightarrow V \rightarrow W$. However, observing V requires detailed and expensive labels. We propose graph aligning approach that generates rich or detailed labels given normal labels $W$. In this paper we investigate the scenario of domain adaptation from the source domain where we have access to the expensive labels $V$ to the target domain where only normal labels W are available. Focusing on the problem of predicting chemical compound graphs from 2D images the fully mediating layer is represented using the planar embedding of the chemical graph structure we are predicting. The use of a fully mediating layer implies some assumptions on the mechanism of the underlying process. However if the assumptions are correct it should allow the machine learning model to be more interpretable, generalize better and be more data efficient at training time. The empirical results show that, using only 4000 data points, we obtain up to 4x improvement of performance after domain adaptation to target domain compared to pretrained model only on the source domain. After domain adaptation, the model is even able to detect atom types that were never seen in the original source domain. Finally, on the Maybridge data set the proposed self-labeling approach reached higher performance than the current state of the art.
Topological Graph Neural Networks
Feb 15, 2021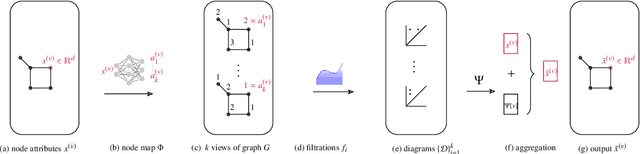
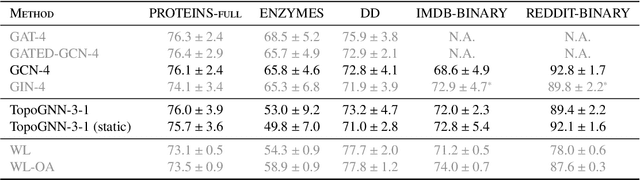
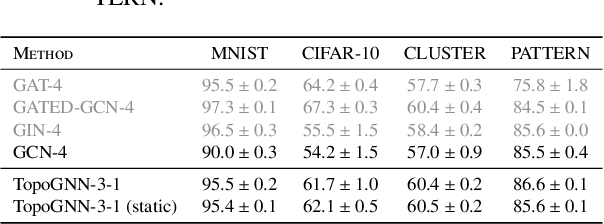
Graph neural networks (GNNs) are a powerful architecture for tackling graph learning tasks, yet have been shown to be oblivious to eminent substructures, such as cycles. We present TOGL, a novel layer that incorporates global topological information of a graph using persistent homology. TOGL can be easily integrated into any type of GNN and is strictly more expressive in terms of the Weisfeiler--Lehman test of isomorphism. Augmenting GNNs with our layer leads to beneficial predictive performance, both on synthetic data sets, which can be trivially classified by humans but not by ordinary GNNs, and on real-world data.
Longitudinal modeling of MS patient trajectories improves predictions of disability progression
Nov 09, 2020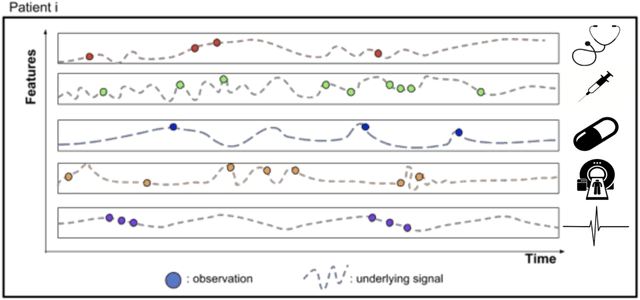
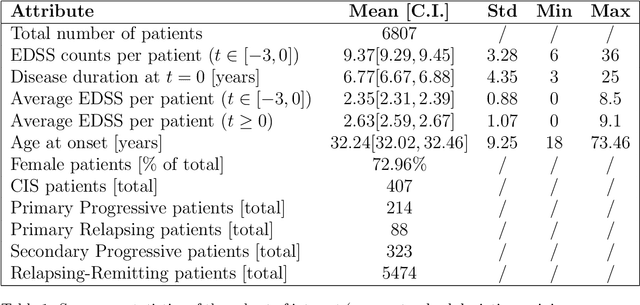
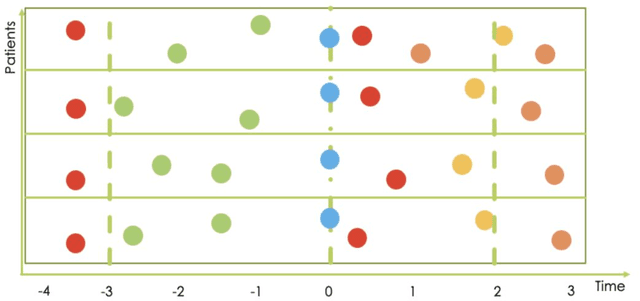

Research in Multiple Sclerosis (MS) has recently focused on extracting knowledge from real-world clinical data sources. This type of data is more abundant than data produced during clinical trials and potentially more informative about real-world clinical practice. However, this comes at the cost of less curated and controlled data sets. In this work, we address the task of optimally extracting information from longitudinal patient data in the real-world setting with a special focus on the sporadic sampling problem. Using the MSBase registry, we show that with machine learning methods suited for patient trajectories modeling, such as recurrent neural networks and tensor factorization, we can predict disability progression of patients in a two-year horizon with an ROC-AUC of 0.86, which represents a 33% decrease in the ranking pair error (1-AUC) compared to reference methods using static clinical features. Compared to the models available in the literature, this work uses the most complete patient history for MS disease progression prediction.