 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIndustry-Scale Orchestrated Federated Learning for Drug Discovery
Oct 17, 2022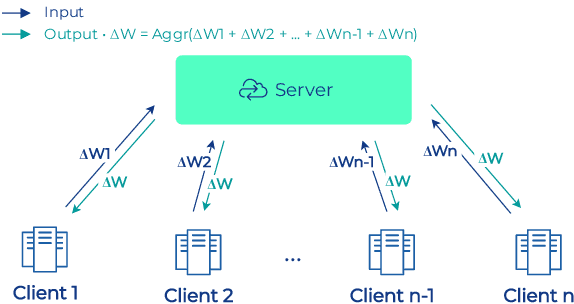
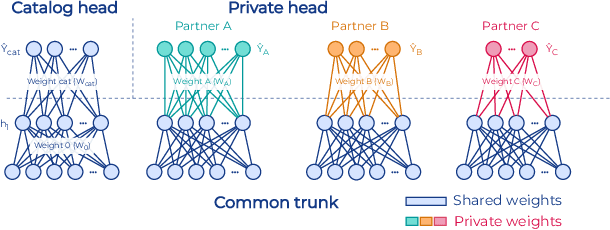
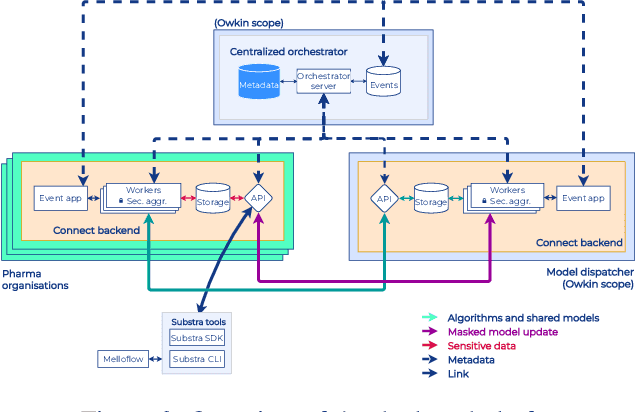
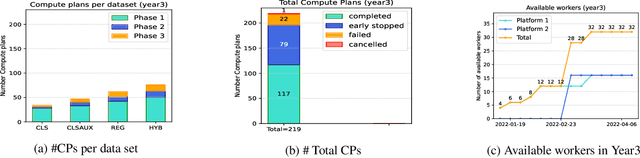
To apply federated learning to drug discovery we developed a novel platform in the context of European Innovative Medicines Initiative (IMI) project MELLODDY (grant n{\deg}831472), which was comprised of 10 pharmaceutical companies, academic research labs, large industrial companies and startups. To the best of our knowledge, The MELLODDY platform was the first industry-scale platform to enable the creation of a global federated model for drug discovery without sharing the confidential data sets of the individual partners. The federated model was trained on the platform by aggregating the gradients of all contributing partners in a cryptographic, secure way following each training iteration. The platform was deployed on an Amazon Web Services (AWS) multi-account architecture running Kubernetes clusters in private subnets. Organisationally, the roles of the different partners were codified as different rights and permissions on the platform and administrated in a decentralized way. The MELLODDY platform generated new scientific discoveries which are described in a companion paper.
SparseChem: Fast and accurate machine learning model for small molecules
Mar 09, 2022SparseChem provides fast and accurate machine learning models for biochemical applications. Especially, the package supports very high-dimensional sparse inputs, e.g., millions of features and millions of compounds. It is possible to train classification, regression and censored regression models, or combination of them from command line. Additionally, the library can be accessed directly from Python. Source code and documentation is freely available under MIT License on GitHub.
Self-Labeling of Fully Mediating Representations by Graph Alignment
Mar 25, 2021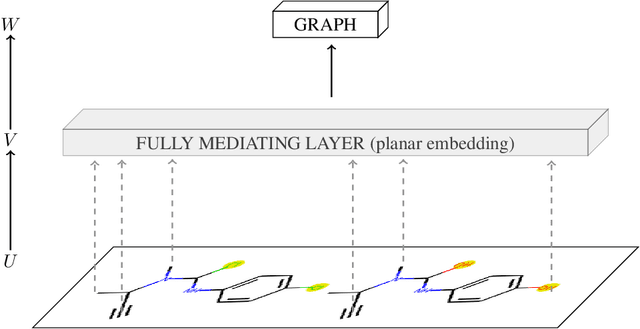

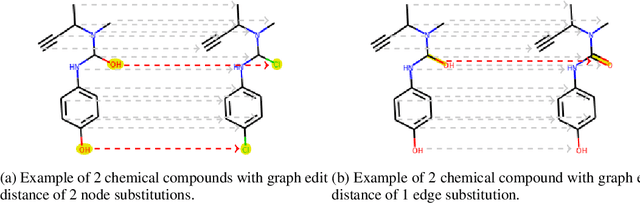
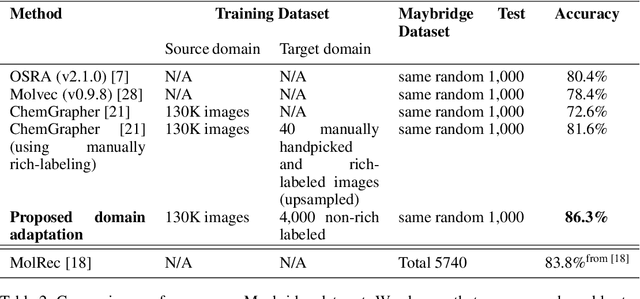
To be able to predict a molecular graph structure ($W$) given a 2D image of a chemical compound ($U$) is a challenging problem in machine learning. We are interested to learn $f: U \rightarrow W$ where we have a fully mediating representation $V$ such that $f$ factors into $U \rightarrow V \rightarrow W$. However, observing V requires detailed and expensive labels. We propose graph aligning approach that generates rich or detailed labels given normal labels $W$. In this paper we investigate the scenario of domain adaptation from the source domain where we have access to the expensive labels $V$ to the target domain where only normal labels W are available. Focusing on the problem of predicting chemical compound graphs from 2D images the fully mediating layer is represented using the planar embedding of the chemical graph structure we are predicting. The use of a fully mediating layer implies some assumptions on the mechanism of the underlying process. However if the assumptions are correct it should allow the machine learning model to be more interpretable, generalize better and be more data efficient at training time. The empirical results show that, using only 4000 data points, we obtain up to 4x improvement of performance after domain adaptation to target domain compared to pretrained model only on the source domain. After domain adaptation, the model is even able to detect atom types that were never seen in the original source domain. Finally, on the Maybridge data set the proposed self-labeling approach reached higher performance than the current state of the art.
Multilevel Gibbs Sampling for Bayesian Regression
Sep 25, 2020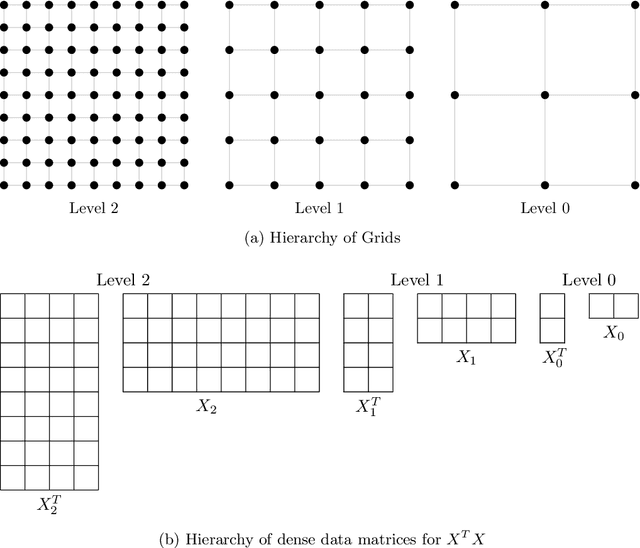



Bayesian regression remains a simple but effective tool based on Bayesian inference techniques. For large-scale applications, with complicated posterior distributions, Markov Chain Monte Carlo methods are applied. To improve the well-known computational burden of Markov Chain Monte Carlo approach for Bayesian regression, we developed a multilevel Gibbs sampler for Bayesian regression of linear mixed models. The level hierarchy of data matrices is created by clustering the features and/or samples of data matrices. Additionally, the use of correlated samples is investigated for variance reduction to improve the convergence of the Markov Chain. Testing on a diverse set of data sets, speed-up is achieved for almost all of them without significant loss in predictive performance.
ChemGrapher: Optical Graph Recognition of Chemical Compounds by Deep Learning
Feb 23, 2020
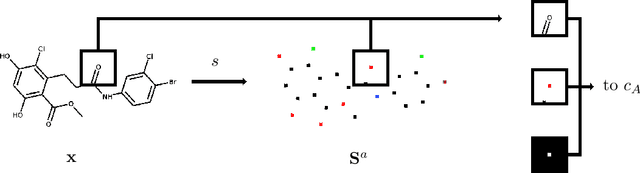
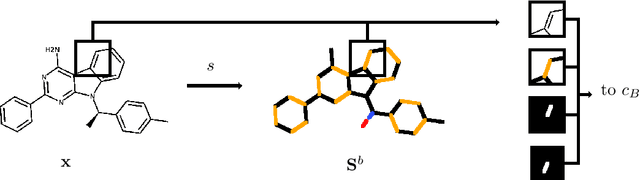

In drug discovery, knowledge of the graph structure of chemical compounds is essential. Many thousands of scientific articles in chemistry and pharmaceutical sciences have investigated chemical compounds, but in cases the details of the structure of these chemical compounds is published only as an images. A tool to analyze these images automatically and convert them into a chemical graph structure would be useful for many applications, such drug discovery. A few such tools are available and they are mostly derived from optical character recognition. However, our evaluation of the performance of those tools reveals that they make often mistakes in detecting the correct bond multiplicity and stereochemical information. In addition, errors sometimes even lead to missing atoms in the resulting graph. In our work, we address these issues by developing a compound recognition method based on machine learning. More specifically, we develop a deep neural network model for optical compound recognition. The deep learning solution presented here consists of a segmentation model, followed by three classification models that predict atom locations, bonds and charges. Furthermore, this model not only predicts the graph structure of the molecule but also produces all information necessary to relate each component of the resulting graph to the source image. This solution is scalable and could rapidly process thousands of images. Finally, we compare empirically the proposed method to a well-established tool and observe significant error reductions.
Graph Informer Networks for Molecules
Jul 25, 2019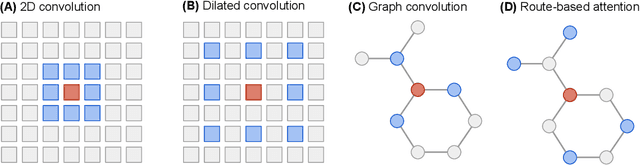
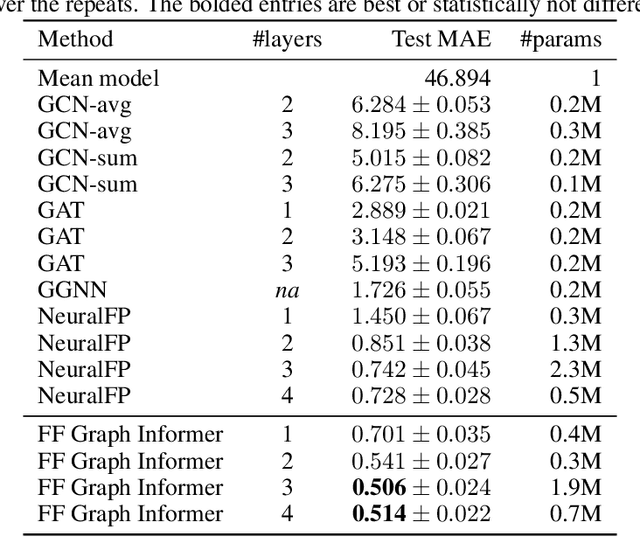
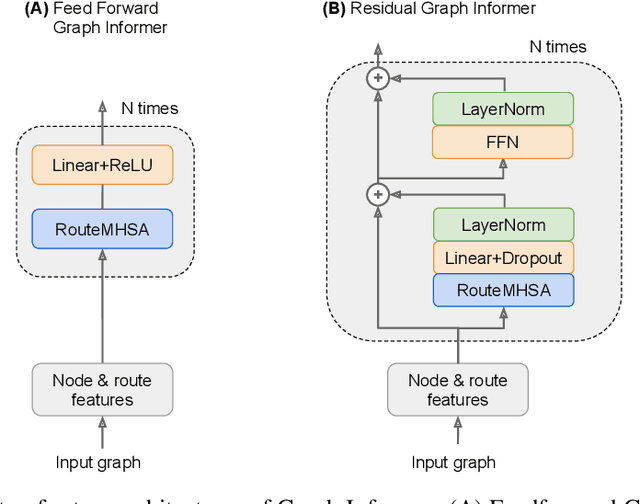

In machine learning, chemical molecules are often represented by sparse high-dimensional vectorial fingerprints. However, a more natural mathematical object for molecule representation is a graph, which is much more challenging to handle from a machine learning perspective. In recent years, several deep learning architectures have been proposed to directly learn from the graph structure of chemical molecules, including graph convolution (Duvenaud et al., 2015) and graph gating networks (Li et al., 2015). Here, we introduce Graph Informer, a route-based multi-head attention mechanism inspired by transformer networks (Vaswani et al., 2017), which incorporates features for node pairs. We show empirically that the proposed method gives significant improvements over existing approaches in prediction tasks for 13C nuclear magnetic resonance spectra and for drug bioactivity. These results indicate that our method is well suited for both node-level and graph-level prediction tasks.
GRU-ODE-Bayes: Continuous modeling of sporadically-observed time series
May 29, 2019



Modeling real-world multidimensional time series can be particularly challenging when these are sporadically observed (i.e., sampling is irregular both in time and across dimensions)-such as in the case of clinical patient data. To address these challenges, we propose (1) a continuous-time version of the Gated Recurrent Unit, building upon the recent Neural Ordinary Differential Equations (Chen et al., 2018), and (2) a Bayesian update network that processes the sporadic observations. We bring these two ideas together in our GRU-ODE-Bayes method. We then demonstrate that the proposed method encodes a continuity prior for the latent process and that it can exactly represent the Fokker-Planck dynamics of complex processes driven by a multidimensional stochastic differential equation. Additionally, empirical evaluation shows that our method outperforms the state of the art on both synthetic data and real-world data with applications in healthcare and climate forecast. What is more, the continuity prior is shown to be well suited for low number of samples settings.
Deep Ensemble Tensor Factorization for Longitudinal Patient Trajectories Classification
Nov 28, 2018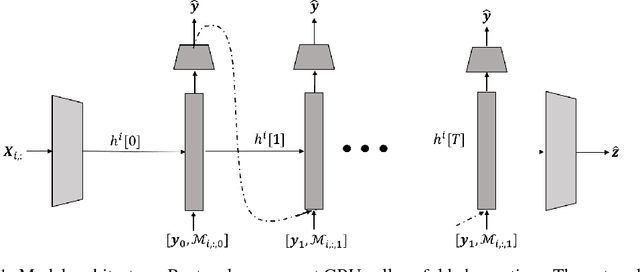
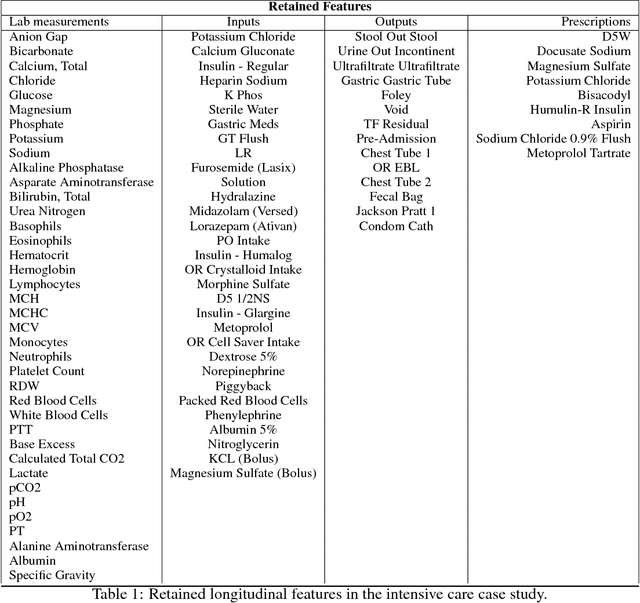
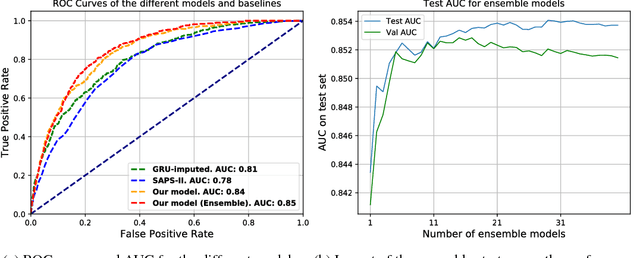
We present a generative approach to classify scarcely observed longitudinal patient trajectories. The available time series are represented as tensors and factorized using generative deep recurrent neural networks. The learned factors represent the patient data in a compact way and can then be used in a downstream classification task. For more robustness and accuracy in the predictions, we used an ensemble of those deep generative models to mimic Bayesian posterior sampling. We illustrate the performance of our architecture on an intensive-care case study of in-hospital mortality prediction with 96 longitudinal measurement types measured across the first 48-hour from admission. Our combination of generative and ensemble strategies achieves an AUC of over 0.85, and outperforms the SAPS-II mortality score and GRU baselines.
Fast semi-supervised discriminant analysis for binary classification of large data-sets
Mar 01, 2018


High-dimensional data requires scalable algorithms. We propose and analyze three scalable and related algorithms for semi-supervised discriminant analysis (SDA). These methods are based on Krylov subspace methods which exploit the data sparsity and the shift-invariance of Krylov subspaces. In addition, the problem definition was improved by adding centralization to the semi-supervised setting. The proposed methods are evaluated on a industry-scale data set from a pharmaceutical company to predict compound activity on target proteins. The results show that SDA achieves good predictive performance and our methods only require a few seconds, significantly improving computation time on previous state of the art.
Macau: Scalable Bayesian Multi-relational Factorization with Side Information using MCMC
Dec 17, 2015

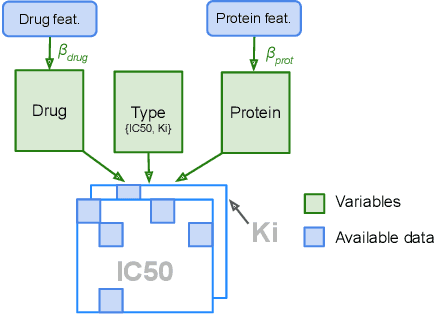
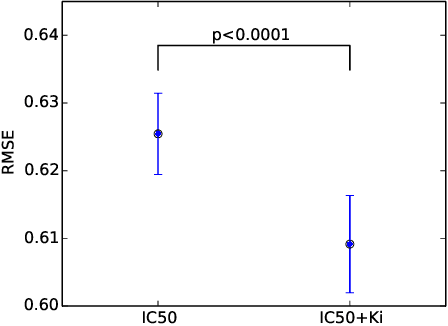
We propose Macau, a powerful and flexible Bayesian factorization method for heterogeneous data. Our model can factorize any set of entities and relations that can be represented by a relational model, including tensors and also multiple relations for each entity. Macau can also incorporate side information, specifically entity and relation features, which are crucial for predicting sparsely observed relations. Macau scales to millions of entity instances, hundred millions of observations, and sparse entity features with millions of dimensions. To achieve the scale up, we specially designed sampling procedure for entity and relation features that relies primarily on noise injection in linear regressions. We show performance and advanced features of Macau in a set of experiments, including challenging drug-protein activity prediction task.