 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Informer Networks for Molecules
Paper and Code
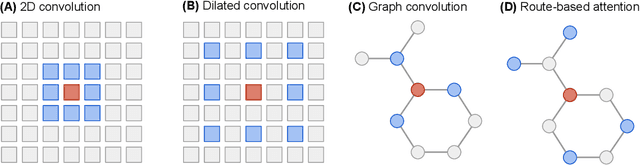
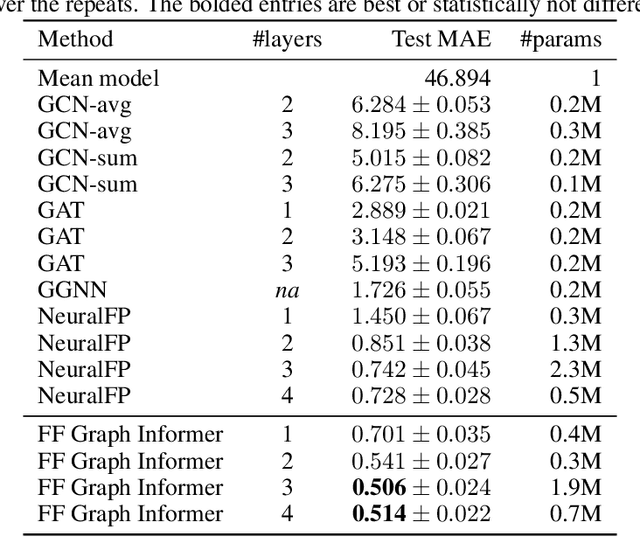
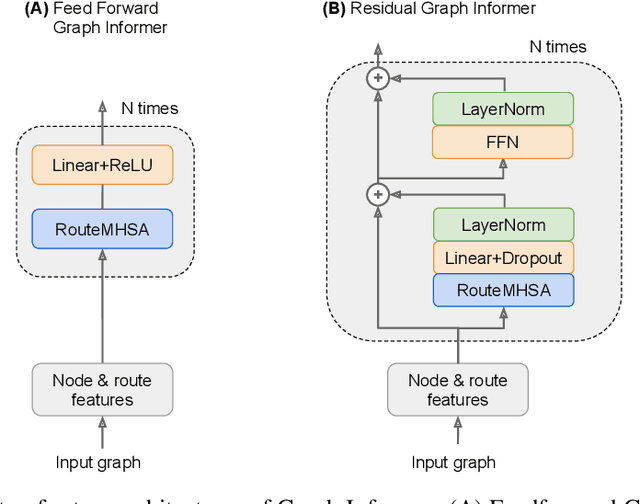

In machine learning, chemical molecules are often represented by sparse high-dimensional vectorial fingerprints. However, a more natural mathematical object for molecule representation is a graph, which is much more challenging to handle from a machine learning perspective. In recent years, several deep learning architectures have been proposed to directly learn from the graph structure of chemical molecules, including graph convolution (Duvenaud et al., 2015) and graph gating networks (Li et al., 2015). Here, we introduce Graph Informer, a route-based multi-head attention mechanism inspired by transformer networks (Vaswani et al., 2017), which incorporates features for node pairs. We show empirically that the proposed method gives significant improvements over existing approaches in prediction tasks for 13C nuclear magnetic resonance spectra and for drug bioactivity. These results indicate that our method is well suited for both node-level and graph-level prediction tasks.