 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTemporal Distribution Shift in Real-World Pharmaceutical Data: Implications for Uncertainty Quantification in QSAR Models
Feb 06, 2025



The estimation of uncertainties associated with predictions from quantitative structure-activity relationship (QSAR) models can accelerate the drug discovery process by identifying promising experiments and allowing an efficient allocation of resources. Several computational tools exist that estimate the predictive uncertainty in machine learning models. However, deviations from the i.i.d. setting have been shown to impair the performance of these uncertainty quantification methods. We use a real-world pharmaceutical dataset to address the pressing need for a comprehensive, large-scale evaluation of uncertainty estimation methods in the context of realistic distribution shifts over time. We investigate the performance of several uncertainty estimation methods, including ensemble-based and Bayesian approaches. Furthermore, we use this real-world setting to systematically assess the distribution shifts in label and descriptor space and their impact on the capability of the uncertainty estimation methods. Our study reveals significant shifts over time in both label and descriptor space and a clear connection between the magnitude of the shift and the nature of the assay. Moreover, we show that pronounced distribution shifts impair the performance of popular uncertainty estimation methods used in QSAR models. This work highlights the challenges of identifying uncertainty quantification methods that remain reliable under distribution shifts introduced by real-world data.
Enhancing Uncertainty Quantification in Drug Discovery with Censored Regression Labels
Sep 06, 2024



In the early stages of drug discovery, decisions regarding which experiments to pursue can be influenced by computational models. These decisions are critical due to the time-consuming and expensive nature of the experiments. Therefore, it is becoming essential to accurately quantify the uncertainty in machine learning predictions, such that resources can be used optimally and trust in the models improves. While computational methods for drug discovery often suffer from limited data and sparse experimental observations, additional information can exist in the form of censored labels that provide thresholds rather than precise values of observations. However, the standard approaches that quantify uncertainty in machine learning cannot fully utilize censored labels. In this work, we adapt ensemble-based, Bayesian, and Gaussian models with tools to learn from censored labels by using the Tobit model from survival analysis. Our results demonstrate that despite the partial information available in censored labels, they are essential to accurately and reliably model the real pharmaceutical setting.
Achieving Well-Informed Decision-Making in Drug Discovery: A Comprehensive Calibration Study using Neural Network-Based Structure-Activity Models
Jul 19, 2024



In the drug discovery process, where experiments can be costly and time-consuming, computational models that predict drug-target interactions are valuable tools to accelerate the development of new therapeutic agents. Estimating the uncertainty inherent in these neural network predictions provides valuable information that facilitates optimal decision-making when risk assessment is crucial. However, such models can be poorly calibrated, which results in unreliable uncertainty estimates that do not reflect the true predictive uncertainty. In this study, we compare different metrics, including accuracy and calibration scores, used for model hyperparameter tuning to investigate which model selection strategy achieves well-calibrated models. Furthermore, we propose to use a computationally efficient Bayesian uncertainty estimation method named Bayesian Linear Probing (BLP), which generates Hamiltonian Monte Carlo (HMC) trajectories to obtain samples for the parameters of a Bayesian Logistic Regression fitted to the hidden layer of the baseline neural network. We report that BLP improves model calibration and achieves the performance of common uncertainty quantification methods by combining the benefits of uncertainty estimation and probability calibration methods. Finally, we show that combining post hoc calibration method with well-performing uncertainty quantification approaches can boost model accuracy and calibration.
Atom-Level Optical Chemical Structure Recognition with Limited Supervision
Apr 02, 2024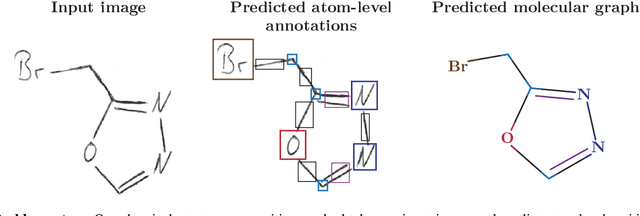


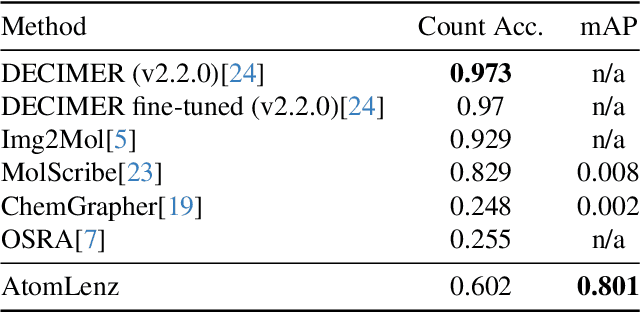
Identifying the chemical structure from a graphical representation, or image, of a molecule is a challenging pattern recognition task that would greatly benefit drug development. Yet, existing methods for chemical structure recognition do not typically generalize well, and show diminished effectiveness when confronted with domains where data is sparse, or costly to generate, such as hand-drawn molecule images. To address this limitation, we propose a new chemical structure recognition tool that delivers state-of-the-art performance and can adapt to new domains with a limited number of data samples and supervision. Unlike previous approaches, our method provides atom-level localization, and can therefore segment the image into the different atoms and bonds. Our model is the first model to perform OCSR with atom-level entity detection with only SMILES supervision. Through rigorous and extensive benchmarking, we demonstrate the preeminence of our chemical structure recognition approach in terms of data efficiency, accuracy, and atom-level entity prediction.
Weakly Supervised Knowledge Transfer with Probabilistic Logical Reasoning for Object Detection
Mar 09, 2023



Training object detection models usually requires instance-level annotations, such as the positions and labels of all objects present in each image. Such supervision is unfortunately not always available and, more often, only image-level information is provided, also known as weak supervision. Recent works have addressed this limitation by leveraging knowledge from a richly annotated domain. However, the scope of weak supervision supported by these approaches has been very restrictive, preventing them to use all available information. In this work, we propose ProbKT, a framework based on probabilistic logical reasoning that allows to train object detection models with arbitrary types of weak supervision. We empirically show on different datasets that using all available information is beneficial as our ProbKT leads to significant improvement on target domain and better generalization compared to existing baselines. We also showcase the ability of our approach to handle complex logic statements as supervision signal.
Collaborative Drug Discovery: Inference-level Data Protection Perspective
May 13, 2022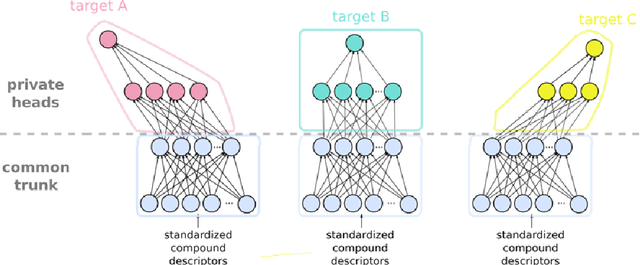
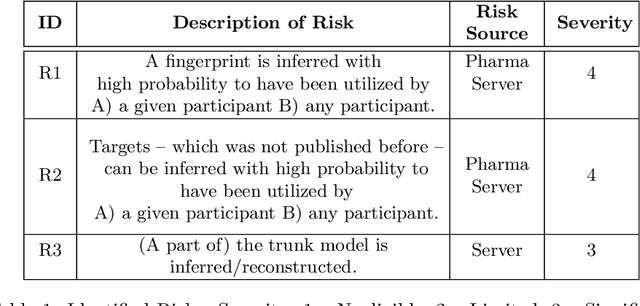
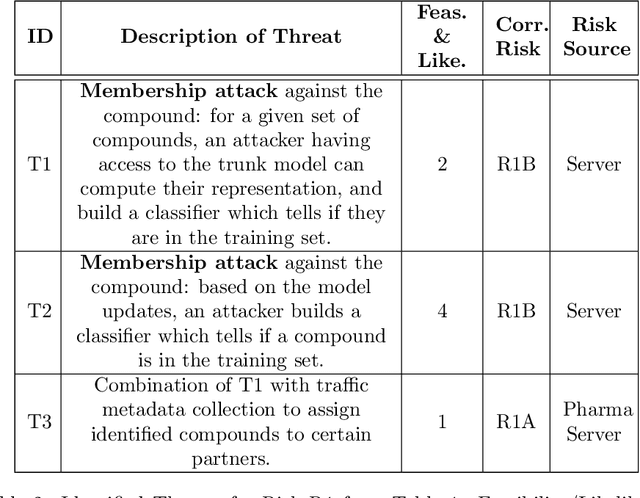
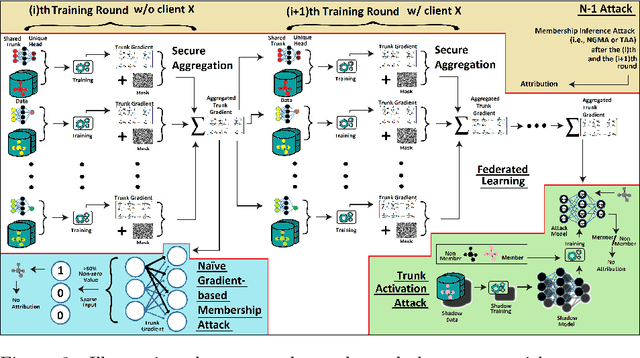
Pharmaceutical industry can better leverage its data assets to virtualize drug discovery through a collaborative machine learning platform. On the other hand, there are non-negligible risks stemming from the unintended leakage of participants' training data, hence, it is essential for such a platform to be secure and privacy-preserving. This paper describes a privacy risk assessment for collaborative modeling in the preclinical phase of drug discovery to accelerate the selection of promising drug candidates. After a short taxonomy of state-of-the-art inference attacks we adopt and customize several to the underlying scenario. Finally we describe and experiments with a handful of relevant privacy protection techniques to mitigate such attacks.
SparseChem: Fast and accurate machine learning model for small molecules
Mar 09, 2022SparseChem provides fast and accurate machine learning models for biochemical applications. Especially, the package supports very high-dimensional sparse inputs, e.g., millions of features and millions of compounds. It is possible to train classification, regression and censored regression models, or combination of them from command line. Additionally, the library can be accessed directly from Python. Source code and documentation is freely available under MIT License on GitHub.
Self-Labeling of Fully Mediating Representations by Graph Alignment
Mar 25, 2021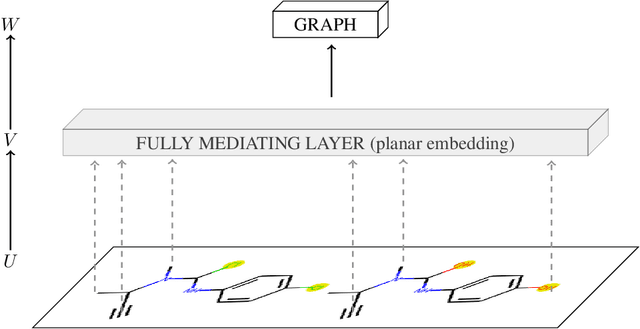

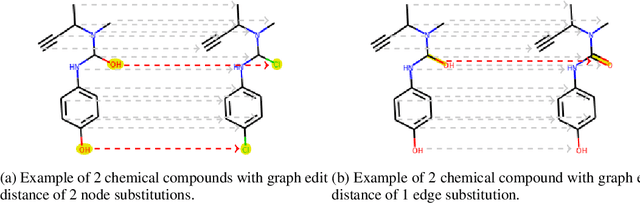
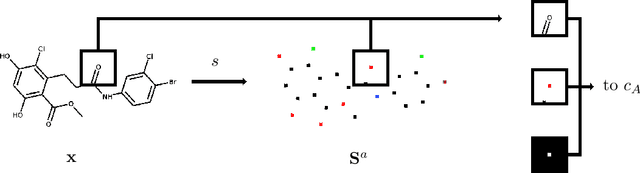
To be able to predict a molecular graph structure ($W$) given a 2D image of a chemical compound ($U$) is a challenging problem in machine learning. We are interested to learn $f: U \rightarrow W$ where we have a fully mediating representation $V$ such that $f$ factors into $U \rightarrow V \rightarrow W$. However, observing V requires detailed and expensive labels. We propose graph aligning approach that generates rich or detailed labels given normal labels $W$. In this paper we investigate the scenario of domain adaptation from the source domain where we have access to the expensive labels $V$ to the target domain where only normal labels W are available. Focusing on the problem of predicting chemical compound graphs from 2D images the fully mediating layer is represented using the planar embedding of the chemical graph structure we are predicting. The use of a fully mediating layer implies some assumptions on the mechanism of the underlying process. However if the assumptions are correct it should allow the machine learning model to be more interpretable, generalize better and be more data efficient at training time. The empirical results show that, using only 4000 data points, we obtain up to 4x improvement of performance after domain adaptation to target domain compared to pretrained model only on the source domain. After domain adaptation, the model is even able to detect atom types that were never seen in the original source domain. Finally, on the Maybridge data set the proposed self-labeling approach reached higher performance than the current state of the art.
Multilevel Gibbs Sampling for Bayesian Regression
Sep 25, 2020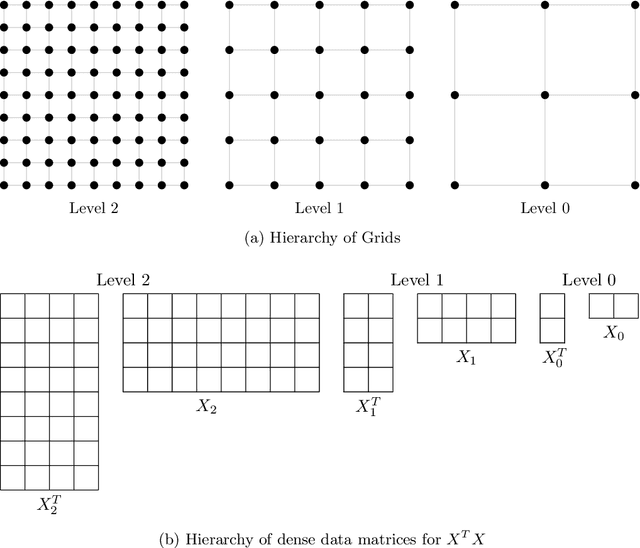



Bayesian regression remains a simple but effective tool based on Bayesian inference techniques. For large-scale applications, with complicated posterior distributions, Markov Chain Monte Carlo methods are applied. To improve the well-known computational burden of Markov Chain Monte Carlo approach for Bayesian regression, we developed a multilevel Gibbs sampler for Bayesian regression of linear mixed models. The level hierarchy of data matrices is created by clustering the features and/or samples of data matrices. Additionally, the use of correlated samples is investigated for variance reduction to improve the convergence of the Markov Chain. Testing on a diverse set of data sets, speed-up is achieved for almost all of them without significant loss in predictive performance.
ChemGrapher: Optical Graph Recognition of Chemical Compounds by Deep Learning
Feb 23, 2020
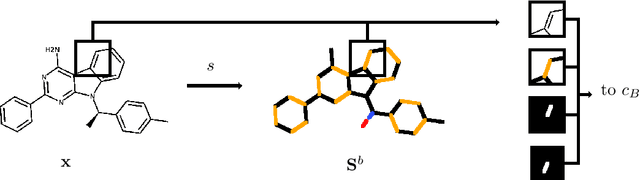

In drug discovery, knowledge of the graph structure of chemical compounds is essential. Many thousands of scientific articles in chemistry and pharmaceutical sciences have investigated chemical compounds, but in cases the details of the structure of these chemical compounds is published only as an images. A tool to analyze these images automatically and convert them into a chemical graph structure would be useful for many applications, such drug discovery. A few such tools are available and they are mostly derived from optical character recognition. However, our evaluation of the performance of those tools reveals that they make often mistakes in detecting the correct bond multiplicity and stereochemical information. In addition, errors sometimes even lead to missing atoms in the resulting graph. In our work, we address these issues by developing a compound recognition method based on machine learning. More specifically, we develop a deep neural network model for optical compound recognition. The deep learning solution presented here consists of a segmentation model, followed by three classification models that predict atom locations, bonds and charges. Furthermore, this model not only predicts the graph structure of the molecule but also produces all information necessary to relate each component of the resulting graph to the source image. This solution is scalable and could rapidly process thousands of images. Finally, we compare empirically the proposed method to a well-established tool and observe significant error reductions.