 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePublishing Neural Networks in Drug Discovery Might Compromise Training Data Privacy
Oct 22, 2024



This study investigates the risks of exposing confidential chemical structures when machine learning models trained on these structures are made publicly available. We use membership inference attacks, a common method to assess privacy that is largely unexplored in the context of drug discovery, to examine neural networks for molecular property prediction in a black-box setting. Our results reveal significant privacy risks across all evaluated datasets and neural network architectures. Combining multiple attacks increases these risks. Molecules from minority classes, often the most valuable in drug discovery, are particularly vulnerable. We also found that representing molecules as graphs and using message-passing neural networks may mitigate these risks. We provide a framework to assess privacy risks of classification models and molecular representations. Our findings highlight the need for careful consideration when sharing neural networks trained on proprietary chemical structures, informing organisations and researchers about the trade-offs between data confidentiality and model openness.
Be aware of overfitting by hyperparameter optimization!
Jul 30, 2024Hyperparameter optimization is very frequently employed in machine learning. However, an optimization of a large space of parameters could result in overfitting of models. In recent studies on solubility prediction the authors collected seven thermodynamic and kinetic solubility datasets from different data sources. They used state-of-the-art graph-based methods and compared models developed for each dataset using different data cleaning protocols and hyperparameter optimization. In our study we showed that hyperparameter optimization did not always result in better models, possibly due to overfitting when using the same statistical measures. Similar results could be calculated using pre-set hyperparameters, reducing the computational effort by around 10,000 times. We also extended the previous analysis by adding a representation learning method based on Natural Language Processing of smiles called Transformer CNN. We show that across all analyzed sets using exactly the same protocol, Transformer CNN provided better results than graph-based methods for 26 out of 28 pairwise comparisons by using only a tiny fraction of time as compared to other methods. Last but not least we stressed the importance of comparing calculation results using exactly the same statistical measures.
Beyond Chemical 1D knowledge using Transformers
Oct 07, 2020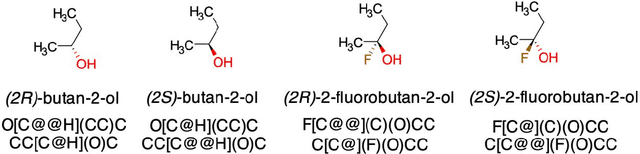


In the present paper we evaluated efficiency of the recent Transformer-CNN models to predict target properties based on the augmented stereochemical SMILES. We selected a well-known Cliff activity dataset as well as a Dipole moment dataset and compared the effect of three representations for R/S stereochemistry in SMILES. The considered representations were SMILES without stereochemistry (noChiSMI), classical relative stereochemistry encoding (RelChiSMI) and an alternative version with absolute stereochemistry encoding (AbsChiSMI). The inclusion of R/S in SMILES representation allowed simplify the assignment of the respective information based on SMILES representation, but did not always show advantages on regression or classification tasks. Interestingly, we did not see degradation of the performance of Transformer-CNN models when the stereochemical information was not present in SMILES. Moreover, these models showed higher or similar performance compared to descriptor-based models based on 3D structures. These observations are an important step in NLP modeling of 3D chemical tasks. An open challenge remains whether Transformer-CNN can efficiently embed 3D knowledge from SMILES input and whether a better representation could further increase the accuracy of this approach.
Augmented Transformer Achieves 97% and 85% for Top5 Prediction of Direct and Classical Retro-Synthesis
Mar 05, 2020

We investigated the effect of different augmentation scenarios on predicting (retro)synthesis of chemical compounds using SMILES representation. We showed that augmentation of not only input sequences but also, importantly, of the target data eliminated the effect of data memorization by neural networks and improved their generalization performance for prediction of new sequences. The Top-5 accuracy was 85.4% for the prediction of the largest fragment (thus identifying principal transformation for classical retro-synthesis) for USPTO-50k test dataset and was achieved by a combination of SMILES augmentation and beam search. The same approach also outperformed best published results for prediction of direct reactions from the USPTO-MIT test set. Our model achieved 90.4% Top-1 and 96.5% Top-5 accuracy for its most challenging mixed set and 97% Top-5 accuracy for the USPTO-MIT separated set. The appearance frequency of the most abundantly generated SMILES was well correlated with the prediction outcome and can be used as a measure of the quality of reaction prediction.
Multitask Learning On Graph Neural Networks Applied To Molecular Property Predictions
Oct 30, 2019
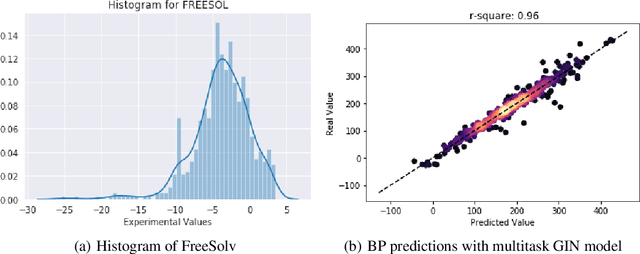
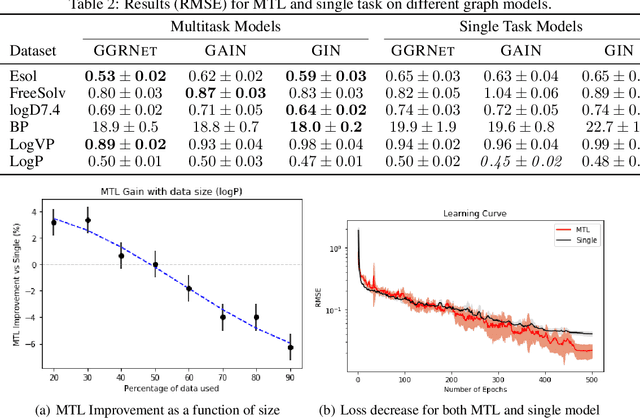
Prediction of molecular properties, including physico-chemical properties, is a challenging task in chemistry. Herein we present a new state-of-the-art multitask prediction method based on existing graph neural network models. We have used different architectures for our models and the results clearly demonstrate that multitask learning can improve model performance. Additionally, a significant reduction of variance in the models has been observed. Most importantly, datasets with a small amount of data points reach better results without the need of augmentation.
Transformer-CNN: Fast and Reliable tool for QSAR
Oct 21, 2019
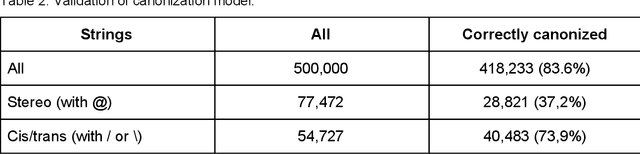

We present SMILES-embeddings derived from internal encoder state of a Transformer[1] model trained to canonize SMILES as a Seq2Seq problem. Using CharNN[2] architecture upon the embeddings results in a higher quality QSAR/QSPR models on diverse benchmark datasets including regression and classification tasks. The proposed Transformer-CNN method uses SMILES augmentation for training and inference, and thus the prognosis grounds on an internal consensus. Both the augmentation and transfer learning based on embedding allows the method to provide good results for small datasets. We discuss the reasons for such effectiveness and draft future directions for the development of the method. The source code and the embeddings are available on https://github.com/bigchem/transformer-cnn, whereas the OCHEM[3] environment (https://ochem.eu) hosts its on-line implementation.
GEN: Highly Efficient SMILES Explorer Using Autodidactic Generative Examination Networks
Sep 10, 2019



Recurrent neural networks have been widely used to generate millions of de novo molecules in a known chemical space. These deep generative models are typically setup with LSTM or GRU units and trained with canonical SMILEs. In this study, we introduce a new robust architecture, Generative Examination Networks GEN, based on bidirectional RNNs with concatenated sub-models to learn and generate molecular SMILES with a trained target space. GENs autonomously learn the target space in a few epochs while being subjected to an independent online examination mechanism to measure the quality of the generated set. Here we have used online statistical quality control (SQC) on the percentage of valid molecules SMILES as an examination measure to select the earliest available stable model weights. Very high levels of valid SMILES (95-98%) can be generated using multiple parallel encoding layers in combination with SMILES augmentation using unrestricted SMILES randomization. Our architecture combines an excellent novelty rate (85-90%) while generating SMILES with a strong conservation of the property space (95-99%). Our flexible examination mechanism is open to other quality criteria.
Synergy Effect between Convolutional Neural Networks and the Multiplicity of SMILES for Improvement of Molecular Prediction
Dec 11, 2018



In our study, we demonstrate the synergy effect between convolutional neural networks and the multiplicity of SMILES. The model we propose, the so-called Convolutional Neural Fingerprint (CNF) model, reaches the accuracy of traditional descriptors such as Dragon (Mauri et al. [22]), RDKit (Landrum [18]), CDK2 (Willighagen et al. [43]) and PyDescriptor (Masand and Rastija [20]). Moreover the CNF model generally performs better than highly fine-tuned traditional descriptors, especially on small data sets, which is of great interest for the chemical field where data sets are generally small due to experimental costs, the availability of molecules or accessibility to private databases. We evaluate the CNF model along with SMILES augmentation during both training and testing. To the best of our knowledge, this is the first time that such a methodology is presented. We show that using the multiplicity of SMILES during training acts as a regulariser and therefore avoids overfitting and can be seen as ensemble learning when considered for testing.