 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGEN: Highly Efficient SMILES Explorer Using Autodidactic Generative Examination Networks
Sep 10, 2019



Recurrent neural networks have been widely used to generate millions of de novo molecules in a known chemical space. These deep generative models are typically setup with LSTM or GRU units and trained with canonical SMILEs. In this study, we introduce a new robust architecture, Generative Examination Networks GEN, based on bidirectional RNNs with concatenated sub-models to learn and generate molecular SMILES with a trained target space. GENs autonomously learn the target space in a few epochs while being subjected to an independent online examination mechanism to measure the quality of the generated set. Here we have used online statistical quality control (SQC) on the percentage of valid molecules SMILES as an examination measure to select the earliest available stable model weights. Very high levels of valid SMILES (95-98%) can be generated using multiple parallel encoding layers in combination with SMILES augmentation using unrestricted SMILES randomization. Our architecture combines an excellent novelty rate (85-90%) while generating SMILES with a strong conservation of the property space (95-99%). Our flexible examination mechanism is open to other quality criteria.
In silico generation of novel, drug-like chemical matter using the LSTM neural network
Jan 08, 2018
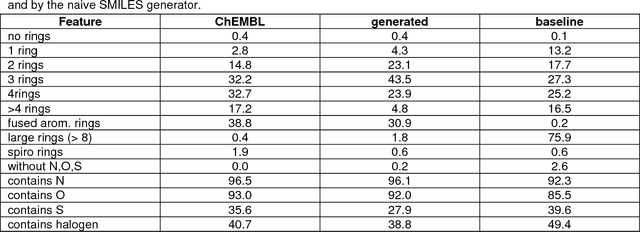
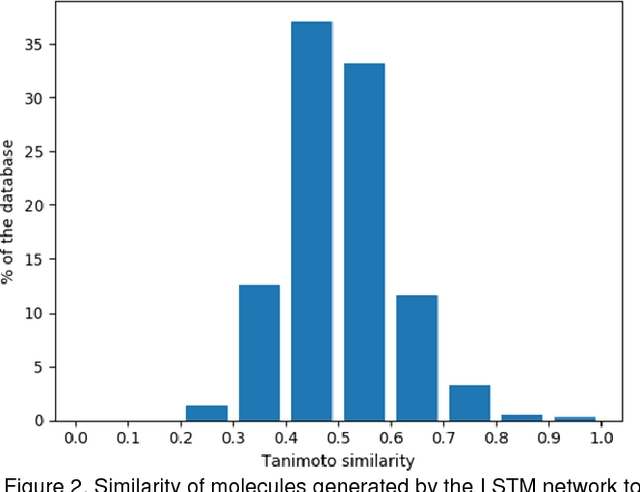
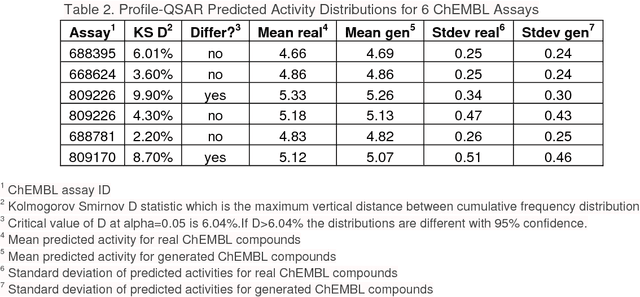
The exploration of novel chemical spaces is one of the most important tasks of cheminformatics when supporting the drug discovery process. Properly designed and trained deep neural networks can provide a viable alternative to brute-force de novo approaches or various other machine-learning techniques for generating novel drug-like molecules. In this article we present a method to generate molecules using a long short-term memory (LSTM) neural network and provide an analysis of the results, including a virtual screening test. Using the network one million drug-like molecules were generated in 2 hours. The molecules are novel, diverse (contain numerous novel chemotypes), have good physicochemical properties and have good synthetic accessibility, even though these qualities were not specific constraints. Although novel, their structural features and functional groups remain closely within the drug-like space defined by the bioactive molecules from ChEMBL. Virtual screening using the profile QSAR approach confirms that the potential of these novel molecules to show bioactivity is comparable to the ChEMBL set from which they were derived. The molecule generator written in Python used in this study is available on request.