 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeParallelizing Linear Recurrent Neural Nets Over Sequence Length
Feb 22, 2018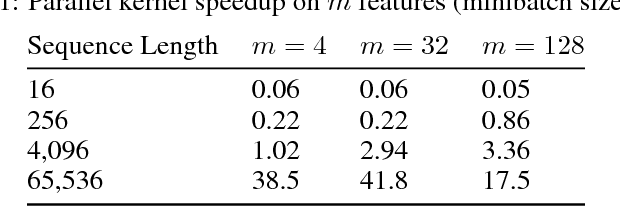
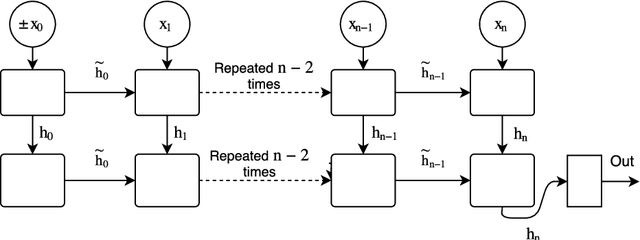
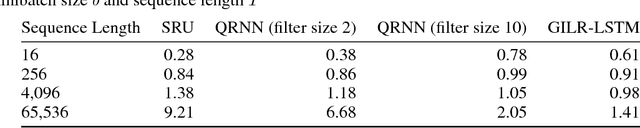
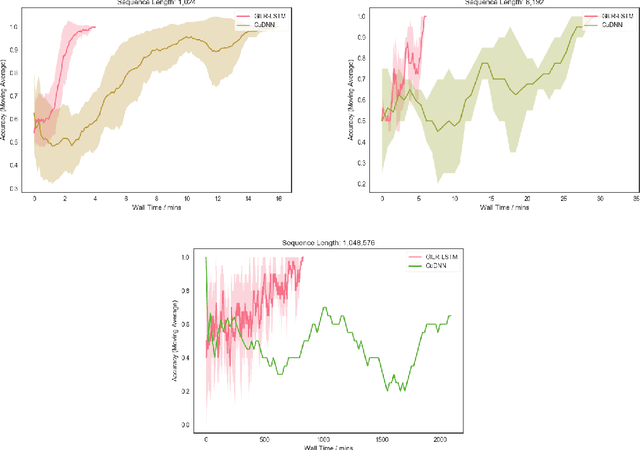
Recurrent neural networks (RNNs) are widely used to model sequential data but their non-linear dependencies between sequence elements prevent parallelizing training over sequence length. We show the training of RNNs with only linear sequential dependencies can be parallelized over the sequence length using the parallel scan algorithm, leading to rapid training on long sequences even with small minibatch size. We develop a parallel linear recurrence CUDA kernel and show that it can be applied to immediately speed up training and inference of several state of the art RNN architectures by up to 9x. We abstract recent work on linear RNNs into a new framework of linear surrogate RNNs and develop a linear surrogate model for the long short-term memory unit, the GILR-LSTM, that utilizes parallel linear recurrence. We extend sequence learning to new extremely long sequence regimes that were previously out of reach by successfully training a GILR-LSTM on a synthetic sequence classification task with a one million timestep dependency.
In silico generation of novel, drug-like chemical matter using the LSTM neural network
Jan 08, 2018
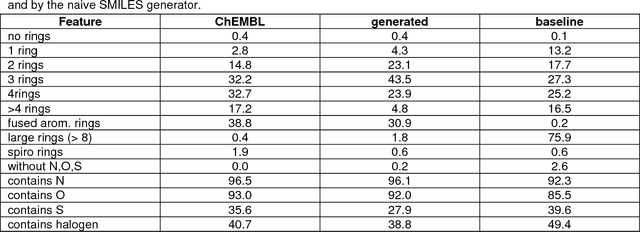
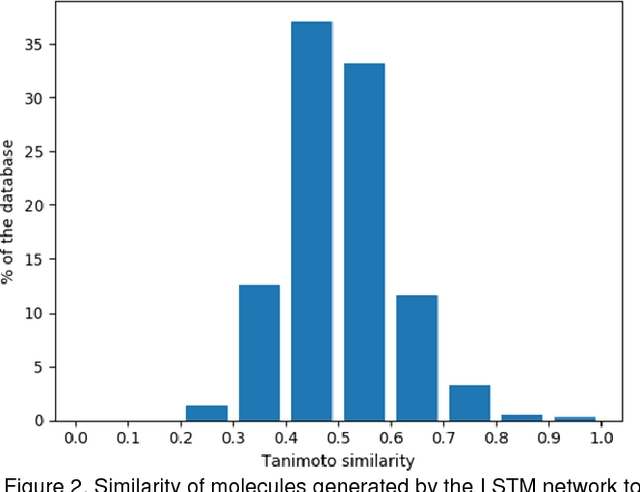
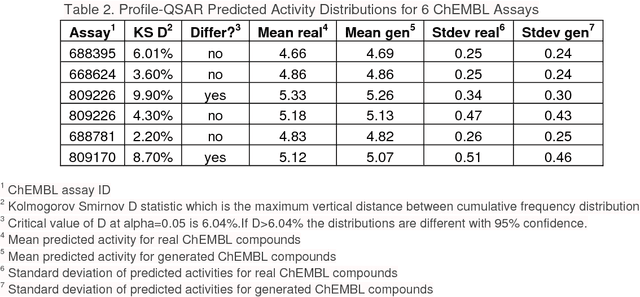
The exploration of novel chemical spaces is one of the most important tasks of cheminformatics when supporting the drug discovery process. Properly designed and trained deep neural networks can provide a viable alternative to brute-force de novo approaches or various other machine-learning techniques for generating novel drug-like molecules. In this article we present a method to generate molecules using a long short-term memory (LSTM) neural network and provide an analysis of the results, including a virtual screening test. Using the network one million drug-like molecules were generated in 2 hours. The molecules are novel, diverse (contain numerous novel chemotypes), have good physicochemical properties and have good synthetic accessibility, even though these qualities were not specific constraints. Although novel, their structural features and functional groups remain closely within the drug-like space defined by the bioactive molecules from ChEMBL. Virtual screening using the profile QSAR approach confirms that the potential of these novel molecules to show bioactivity is comparable to the ChEMBL set from which they were derived. The molecule generator written in Python used in this study is available on request.