Get our free extension to see links to code for papers anywhere online!Free add-on: code for papers everywhere!Free add-on: See code for papers anywhere!
 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultitask Learning On Graph Neural Networks Applied To Molecular Property Predictions
Paper and Code

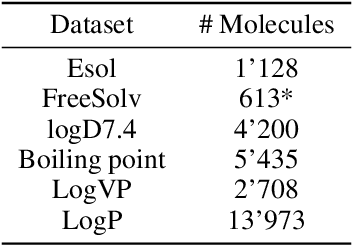
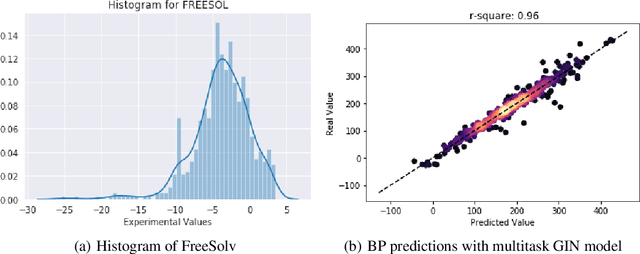
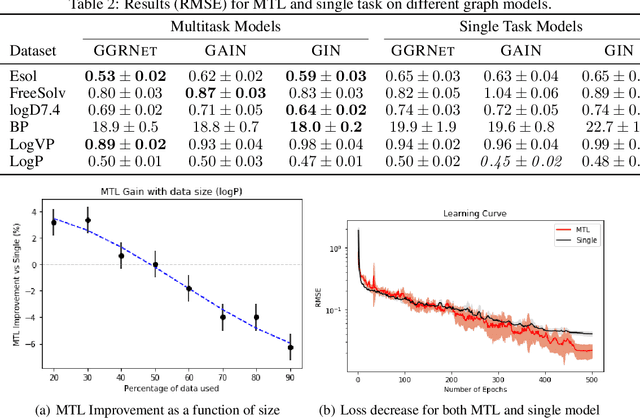
Prediction of molecular properties, including physico-chemical properties, is a challenging task in chemistry. Herein we present a new state-of-the-art multitask prediction method based on existing graph neural network models. We have used different architectures for our models and the results clearly demonstrate that multitask learning can improve model performance. Additionally, a significant reduction of variance in the models has been observed. Most importantly, datasets with a small amount of data points reach better results without the need of augmentation.
View paper on