 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCross-Slice Knowledge Transfer via Masked Multi-Modal Heterogeneous Graph Contrastive Learning for Spatial Gene Expression Inference
Mar 24, 2026While spatial transcriptomics (ST) has advanced our understanding of gene expression in tissue context, its high experimental cost limits its large-scale application. Predicting ST from pathology images is a promising, cost-effective alternative, but existing methods struggle to capture complex cross-slide spatial relationships. To address the challenge, we propose SpaHGC, a multi-modal heterogeneous graph-based model that captures both intra-slice and inter-slice spot-spot relationships from histology images. It integrates local spatial context within the target slide and cross-slide similarities computed from image embeddings extracted by a pathology foundation model. These embeddings enable inter-slice knowledge transfer, and SpaHGC further incorporates Masked Graph Contrastive Learning to enhance feature representation and transfer spatial gene expression knowledge from reference to target slides, enabling it to model complex spatial dependencies and significantly improve prediction accuracy. We conducted comprehensive benchmarking on seven matched histology-ST datasets from different platforms, tissues, and cancer subtypes. The results demonstrate that SpaHGC significantly outperforms the existing nine state-of-the-art methods across all evaluation metrics. Additionally, the predictions are significantly enriched in multiple cancer-related pathways, thereby highlighting its strong biological relevance and application potential.
Adapting a Pre-trained Single-Cell Foundation Model to Spatial Gene Expression Generation from Histology Images
Mar 20, 2026Spatial transcriptomics (ST) enables spot-level in situ expression profiling, but its high cost and limited throughput motivate predicting expression directly from HE-stained histology. Recent advances explore using score- or flow-based generative models to estimate the conditional distribution of gene expression from histology, offering a flexible alternative to deterministic regression approaches. However, most existing generative approaches omit explicit modeling of gene-gene dependencies, undermining biological coherence. Single-cell foundation models (sc-FMs), pre-trained across diverse cell populations, capture these critical gene relationships that histology alone cannot reveal. Yet, applying expression-only sc-FMs to histology-conditioned expression modeling is nontrivial due to the absence of a visual pathway, a mismatch between their pre-training and conditional ST objectives, and the scarcity of mixed-cell ST supervision. To address these challenges, we propose HINGE (HIstology-coNditioned GEneration), which retrofits a pre-trained sc-FM into a conditional expression generator while mostly preserving its learned gene relationships. We achieve this by introducing SoftAdaLN, a lightweight, identity-initialized modulation that injects layer-wise visual context into the backbone, coupled with an expression-space masked diffusion objective and a warm-start curriculum to ensure objective alignment and training stability. Evaluated on three ST datasets, ours outperforms state-of-the-art baselines on mean Pearson correlation and yields more accurate spatial marker expression patterns and higher pairwise co-expression consistency, establishing a practical route to adapt pre-trained sc-FMs for histology-conditioned spatial expression generation.
Supervision-by-Hallucination-and-Transfer: A Weakly-Supervised Approach for Robust and Precise Facial Landmark Detection
Jan 19, 2026High-precision facial landmark detection (FLD) relies on high-resolution deep feature representations. However, low-resolution face images or the compression (via pooling or strided convolution) of originally high-resolution images hinder the learning of such features, thereby reducing FLD accuracy. Moreover, insufficient training data and imprecise annotations further degrade performance. To address these challenges, we propose a weakly-supervised framework called Supervision-by-Hallucination-and-Transfer (SHT) for more robust and precise FLD. SHT contains two novel mutually enhanced modules: Dual Hallucination Learning Network (DHLN) and Facial Pose Transfer Network (FPTN). By incorporating FLD and face hallucination tasks, DHLN is able to learn high-resolution representations with low-resolution inputs for recovering both facial structures and local details and generating more effective landmark heatmaps. Then, by transforming faces from one pose to another, FPTN can further improve landmark heatmaps and faces hallucinated by DHLN for detecting more accurate landmarks. To the best of our knowledge, this is the first study to explore weakly-supervised FLD by integrating face hallucination and facial pose transfer tasks. Experimental results of both face hallucination and FLD demonstrate that our method surpasses state-of-the-art techniques.
FGTBT: Frequency-Guided Task-Balancing Transformer for Unified Facial Landmark Detection
Jan 19, 2026Recently, deep learning based facial landmark detection (FLD) methods have achieved considerable success. However, in challenging scenarios such as large pose variations, illumination changes, and facial expression variations, they still struggle to accurately capture the geometric structure of the face, resulting in performance degradation. Moreover, the limited size and diversity of existing FLD datasets hinder robust model training, leading to reduced detection accuracy. To address these challenges, we propose a Frequency-Guided Task-Balancing Transformer (FGTBT), which enhances facial structure perception through frequency-domain modeling and multi-dataset unified training. Specifically, we propose a novel Fine-Grained Multi-Task Balancing loss (FMB-loss), which moves beyond coarse task-level balancing by assigning weights to individual landmarks based on their occurrence across datasets. This enables more effective unified training and mitigates the issue of inconsistent gradient magnitudes. Additionally, a Frequency-Guided Structure-Aware (FGSA) model is designed to utilize frequency-guided structure injection and regularization to help learn facial structure constraints. Extensive experimental results on popular benchmark datasets demonstrate that the integration of the proposed FMB-loss and FGSA model into our FGTBT framework achieves performance comparable to state-of-the-art methods. The code is available at https://github.com/Xi0ngxinyu/FGTBT.
ClusIR: Towards Cluster-Guided All-in-One Image Restoration
Dec 11, 2025All-in-One Image Restoration (AiOIR) aims to recover high-quality images from diverse degradations within a unified framework. However, existing methods often fail to explicitly model degradation types and struggle to adapt their restoration behavior to complex or mixed degradations. To address these issues, we propose ClusIR, a Cluster-Guided Image Restoration framework that explicitly models degradation semantics through learnable clustering and propagates cluster-aware cues across spatial and frequency domains for adaptive restoration. Specifically, ClusIR comprises two key components: a Probabilistic Cluster-Guided Routing Mechanism (PCGRM) and a Degradation-Aware Frequency Modulation Module (DAFMM). The proposed PCGRM disentangles degradation recognition from expert activation, enabling discriminative degradation perception and stable expert routing. Meanwhile, DAFMM leverages the cluster-guided priors to perform adaptive frequency decomposition and targeted modulation, collaboratively refining structural and textural representations for higher restoration fidelity. The cluster-guided synergy seamlessly bridges semantic cues with frequency-domain modulation, empowering ClusIR to attain remarkable restoration results across a wide range of degradations. Extensive experiments on diverse benchmarks validate that ClusIR reaches competitive performance under several scenarios.
GraphMMP: A Graph Neural Network Model with Mutual Information and Global Fusion for Multimodal Medical Prognosis
Aug 24, 2025In the field of multimodal medical data analysis, leveraging diverse types of data and understanding their hidden relationships continues to be a research focus. The main challenges lie in effectively modeling the complex interactions between heterogeneous data modalities with distinct characteristics while capturing both local and global dependencies across modalities. To address these challenges, this paper presents a two-stage multimodal prognosis model, GraphMMP, which is based on graph neural networks. The proposed model constructs feature graphs using mutual information and features a global fusion module built on Mamba, which significantly boosts prognosis performance. Empirical results show that GraphMMP surpasses existing methods on datasets related to liver prognosis and the METABRIC study, demonstrating its effectiveness in multimodal medical prognosis tasks.
ITCFN: Incomplete Triple-Modal Co-Attention Fusion Network for Mild Cognitive Impairment Conversion Prediction
Jan 20, 2025


Alzheimer's disease (AD) is a common neurodegenerative disease among the elderly. Early prediction and timely intervention of its prodromal stage, mild cognitive impairment (MCI), can decrease the risk of advancing to AD. Combining information from various modalities can significantly improve predictive accuracy. However, challenges such as missing data and heterogeneity across modalities complicate multimodal learning methods as adding more modalities can worsen these issues. Current multimodal fusion techniques often fail to adapt to the complexity of medical data, hindering the ability to identify relationships between modalities. To address these challenges, we propose an innovative multimodal approach for predicting MCI conversion, focusing specifically on the issues of missing positron emission tomography (PET) data and integrating diverse medical information. The proposed incomplete triple-modal MCI conversion prediction network is tailored for this purpose. Through the missing modal generation module, we synthesize the missing PET data from the magnetic resonance imaging and extract features using specifically designed encoders. We also develop a channel aggregation module and a triple-modal co-attention fusion module to reduce feature redundancy and achieve effective multimodal data fusion. Furthermore, we design a loss function to handle missing modality issues and align cross-modal features. These components collectively harness multimodal data to boost network performance. Experimental results on the ADNI1 and ADNI2 datasets show that our method significantly surpasses existing unimodal and other multimodal models. Our code is available at https://github.com/justinhxy/ITFC.
BS-LDM: Effective Bone Suppression in High-Resolution Chest X-Ray Images with Conditional Latent Diffusion Models
Dec 24, 2024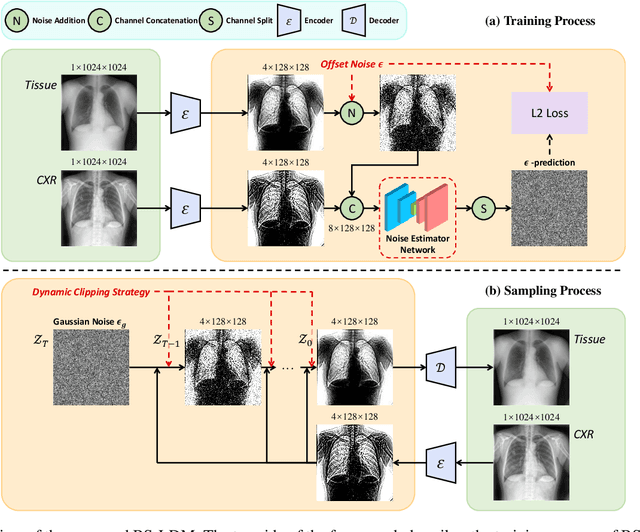
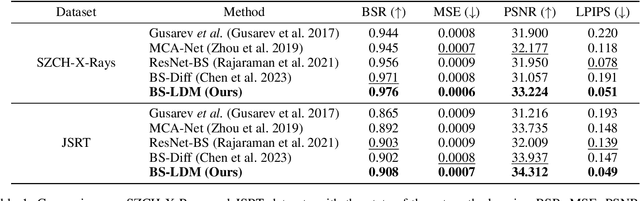
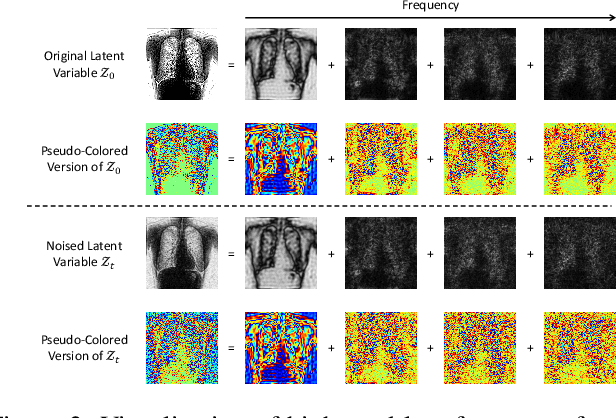
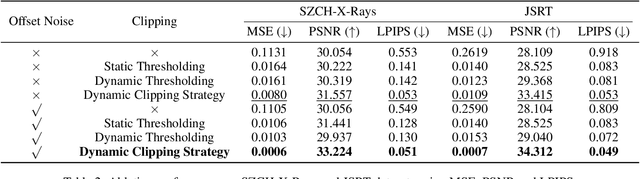
The interference of overlapping bones and pulmonary structures can reduce the effectiveness of Chest X-ray (CXR) examinations. Bone suppression techniques have been developed to improve diagnostic accuracy. Dual-energy subtraction (DES) imaging, a common method for bone suppression, is costly and exposes patients to higher radiation levels. Deep learning-based image generation methods have been proposed as alternatives, however, they often fail to produce high-quality and high-resolution images, resulting in the loss of critical lesion information and texture details. To address these issues, in this paper, we introduce an end-to-end framework for bone suppression in high-resolution CXR images, termed BS-LDM. This framework employs a conditional latent diffusion model to generate high-resolution soft tissue images with fine detail and critical lung pathology by performing bone suppression in the latent space. We implement offset noise during the noise addition phase of the training process to better render low-frequency information in soft tissue images. Additionally, we introduce a dynamic clipping strategy during the sampling process to refine pixel intensity in the generated soft tissue images. We compiled a substantial and high-quality bone suppression dataset, SZCH-X-Rays, including high-resolution paired CXR and DES soft tissue images from 818 patients, collected from our partner hospitals. Moreover, we pre-processed 241 pairs of CXR and DES soft tissue images from the JSRT dataset, the largest publicly available dataset. Comprehensive experimental and clinical evaluations demonstrate that BS-LDM exhibits superior bone suppression capabilities, highlighting its significant clinical potential.
Precise Facial Landmark Detection by Dynamic Semantic Aggregation Transformer
Dec 01, 2024



At present, deep neural network methods have played a dominant role in face alignment field. However, they generally use predefined network structures to predict landmarks, which tends to learn general features and leads to mediocre performance, e.g., they perform well on neutral samples but struggle with faces exhibiting large poses or occlusions. Moreover, they cannot effectively deal with semantic gaps and ambiguities among features at different scales, which may hinder them from learning efficient features. To address the above issues, in this paper, we propose a Dynamic Semantic-Aggregation Transformer (DSAT) for more discriminative and representative feature (i.e., specialized feature) learning. Specifically, a Dynamic Semantic-Aware (DSA) model is first proposed to partition samples into subsets and activate the specific pathways for them by estimating the semantic correlations of feature channels, making it possible to learn specialized features from each subset. Then, a novel Dynamic Semantic Specialization (DSS) model is designed to mine the homogeneous information from features at different scales for eliminating the semantic gap and ambiguities and enhancing the representation ability. Finally, by integrating the DSA model and DSS model into our proposed DSAT in both dynamic architecture and dynamic parameter manners, more specialized features can be learned for achieving more precise face alignment. It is interesting to show that harder samples can be handled by activating more feature channels. Extensive experiments on popular face alignment datasets demonstrate that our proposed DSAT outperforms state-of-the-art models in the literature.Our code is available at https://github.com/GERMINO-LiuHe/DSAT.
Pretrained-Guided Conditional Diffusion Models for Microbiome Data Analysis
Aug 10, 2024Emerging evidence indicates that human cancers are intricately linked to human microbiomes, forming an inseparable connection. However, due to limited sample sizes and significant data loss during collection for various reasons, some machine learning methods have been proposed to address the issue of missing data. These methods have not fully utilized the known clinical information of patients to enhance the accuracy of data imputation. Therefore, we introduce mbVDiT, a novel pre-trained conditional diffusion model for microbiome data imputation and denoising, which uses the unmasked data and patient metadata as conditional guidance for imputating missing values. It is also uses VAE to integrate the the other public microbiome datasets to enhance model performance. The results on the microbiome datasets from three different cancer types demonstrate the performance of our methods in comparison with existing methods.