 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePretrained-Guided Conditional Diffusion Models for Microbiome Data Analysis
Aug 10, 2024Emerging evidence indicates that human cancers are intricately linked to human microbiomes, forming an inseparable connection. However, due to limited sample sizes and significant data loss during collection for various reasons, some machine learning methods have been proposed to address the issue of missing data. These methods have not fully utilized the known clinical information of patients to enhance the accuracy of data imputation. Therefore, we introduce mbVDiT, a novel pre-trained conditional diffusion model for microbiome data imputation and denoising, which uses the unmasked data and patient metadata as conditional guidance for imputating missing values. It is also uses VAE to integrate the the other public microbiome datasets to enhance model performance. The results on the microbiome datasets from three different cancer types demonstrate the performance of our methods in comparison with existing methods.
scASDC: Attention Enhanced Structural Deep Clustering for Single-cell RNA-seq Data
Aug 09, 2024



Single-cell RNA sequencing (scRNA-seq) data analysis is pivotal for understanding cellular heterogeneity. However, the high sparsity and complex noise patterns inherent in scRNA-seq data present significant challenges for traditional clustering methods. To address these issues, we propose a deep clustering method, Attention-Enhanced Structural Deep Embedding Graph Clustering (scASDC), which integrates multiple advanced modules to improve clustering accuracy and robustness.Our approach employs a multi-layer graph convolutional network (GCN) to capture high-order structural relationships between cells, termed as the graph autoencoder module. To mitigate the oversmoothing issue in GCNs, we introduce a ZINB-based autoencoder module that extracts content information from the data and learns latent representations of gene expression. These modules are further integrated through an attention fusion mechanism, ensuring effective combination of gene expression and structural information at each layer of the GCN. Additionally, a self-supervised learning module is incorporated to enhance the robustness of the learned embeddings. Extensive experiments demonstrate that scASDC outperforms existing state-of-the-art methods, providing a robust and effective solution for single-cell clustering tasks. Our method paves the way for more accurate and meaningful analysis of single-cell RNA sequencing data, contributing to better understanding of cellular heterogeneity and biological processes. All code and public datasets used in this paper are available at \url{https://github.com/wenwenmin/scASDC} and \url{https://zenodo.org/records/12814320}.
Masked Graph Autoencoders with Contrastive Augmentation for Spatially Resolved Transcriptomics Data
Aug 09, 2024



With the rapid advancement of Spatial Resolved Transcriptomics (SRT) technology, it is now possible to comprehensively measure gene transcription while preserving the spatial context of tissues. Spatial domain identification and gene denoising are key objectives in SRT data analysis. We propose a Contrastively Augmented Masked Graph Autoencoder (STMGAC) to learn low-dimensional latent representations for domain identification. In the latent space, persistent signals for representations are obtained through self-distillation to guide self-supervised matching. At the same time, positive and negative anchor pairs are constructed using triplet learning to augment the discriminative ability. We evaluated the performance of STMGAC on five datasets, achieving results superior to those of existing baseline methods. All code and public datasets used in this paper are available at https://github.com/wenwenmin/STMGAC and https://zenodo.org/records/13253801.
High-Resolution Spatial Transcriptomics from Histology Images using HisToSGE
Jul 30, 2024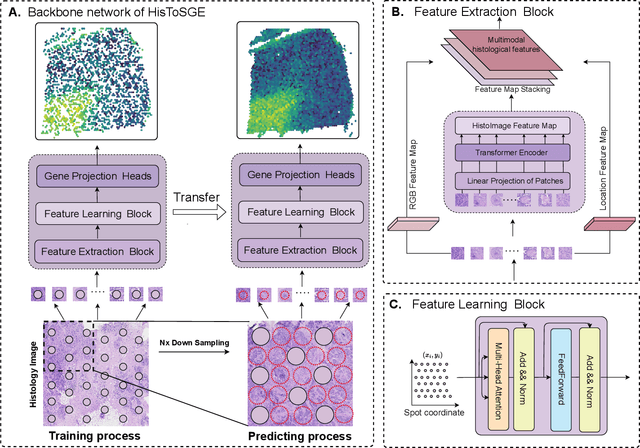
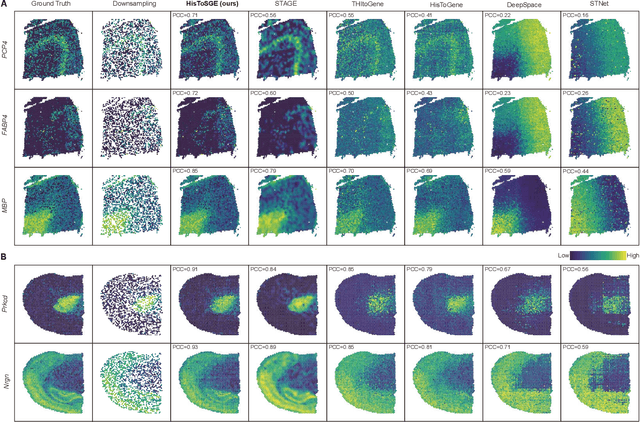
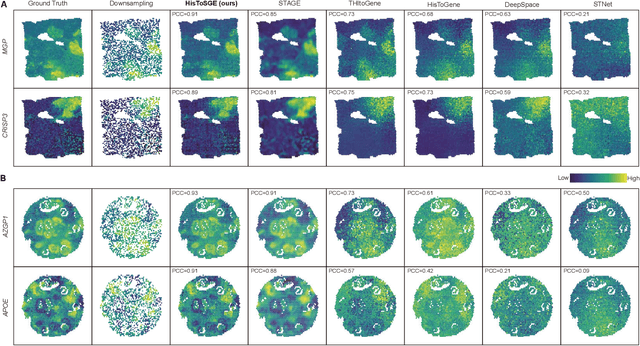
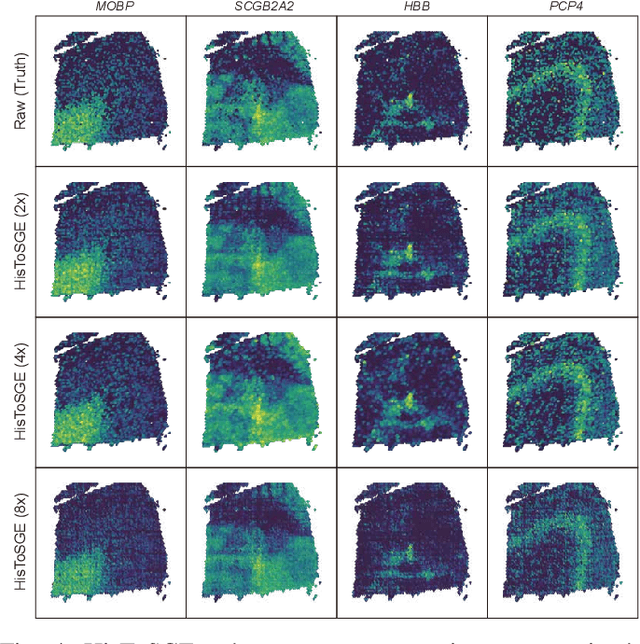
Spatial transcriptomics (ST) is a groundbreaking genomic technology that enables spatial localization analysis of gene expression within tissue sections. However, it is significantly limited by high costs and sparse spatial resolution. An alternative, more cost-effective strategy is to use deep learning methods to predict high-density gene expression profiles from histological images. However, existing methods struggle to capture rich image features effectively or rely on low-dimensional positional coordinates, making it difficult to accurately predict high-resolution gene expression profiles. To address these limitations, we developed HisToSGE, a method that employs a Pathology Image Large Model (PILM) to extract rich image features from histological images and utilizes a feature learning module to robustly generate high-resolution gene expression profiles. We evaluated HisToSGE on four ST datasets, comparing its performance with five state-of-the-art baseline methods. The results demonstrate that HisToSGE excels in generating high-resolution gene expression profiles and performing downstream tasks such as spatial domain identification. All code and public datasets used in this paper are available at https://github.com/wenwenmin/HisToSGE and https://zenodo.org/records/12792163.
SpaDiT: Diffusion Transformer for Spatial Gene Expression Prediction using scRNA-seq
Jul 18, 2024The rapid development of spatial transcriptomics (ST) technologies is revolutionizing our understanding of the spatial organization of biological tissues. Current ST methods, categorized into next-generation sequencing-based (seq-based) and fluorescence in situ hybridization-based (image-based) methods, offer innovative insights into the functional dynamics of biological tissues. However, these methods are limited by their cellular resolution and the quantity of genes they can detect. To address these limitations, we propose SpaDiT, a deep learning method that utilizes a diffusion generative model to integrate scRNA-seq and ST data for the prediction of undetected genes. By employing a Transformer-based diffusion model, SpaDiT not only accurately predicts unknown genes but also effectively generates the spatial structure of ST genes. We have demonstrated the effectiveness of SpaDiT through extensive experiments on both seq-based and image-based ST data. SpaDiT significantly contributes to ST gene prediction methods with its innovative approach. Compared to eight leading baseline methods, SpaDiT achieved state-of-the-art performance across multiple metrics, highlighting its substantial bioinformatics contribution.
stEnTrans: Transformer-based deep learning for spatial transcriptomics enhancement
Jul 11, 2024The spatial location of cells within tissues and organs is crucial for the manifestation of their specific functions.Spatial transcriptomics technology enables comprehensive measurement of the gene expression patterns in tissues while retaining spatial information. However, current popular spatial transcriptomics techniques either have shallow sequencing depth or low resolution. We present stEnTrans, a deep learning method based on Transformer architecture that provides comprehensive predictions for gene expression in unmeasured areas or unexpectedly lost areas and enhances gene expression in original and inputed spots. Utilizing a self-supervised learning approach, stEnTrans establishes proxy tasks on gene expression profile without requiring additional data, mining intrinsic features of the tissues as supervisory information. We evaluate stEnTrans on six datasets and the results indicate superior performance in enhancing spots resolution and predicting gene expression in unmeasured areas compared to other deep learning and traditional interpolation methods. Additionally, Our method also can help the discovery of spatial patterns in Spatial Transcriptomics and enrich to more biologically significant pathways. Our source code is available at https://github.com/shuailinxue/stEnTrans.