 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigh-Resolution Spatial Transcriptomics from Histology Images using HisToSGE
Jul 30, 2024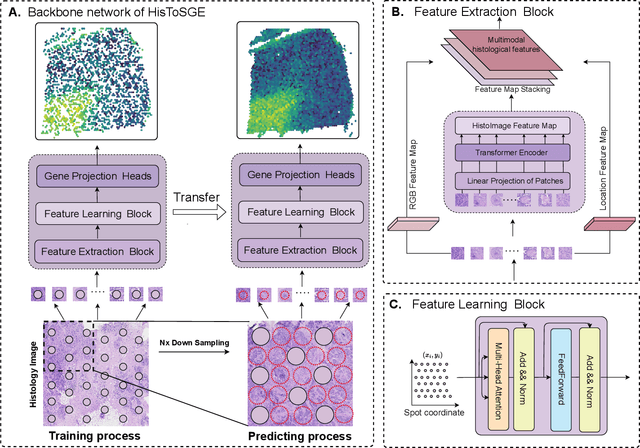
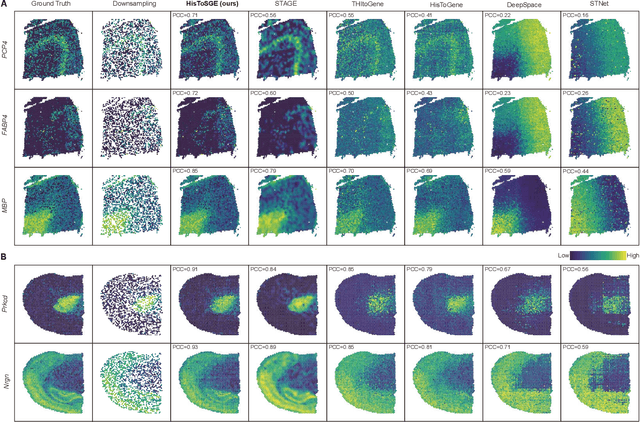
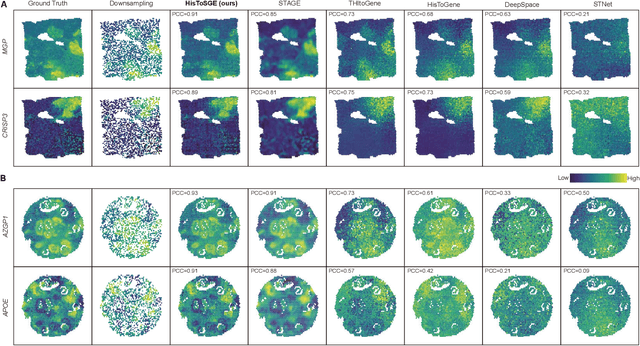
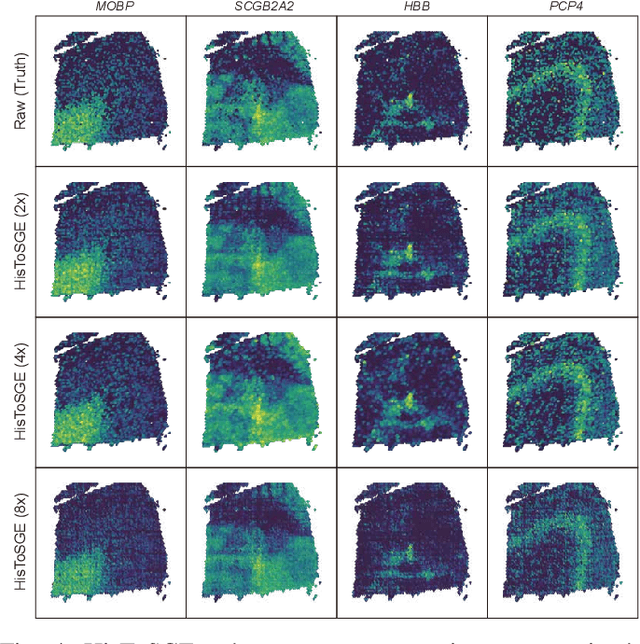
Spatial transcriptomics (ST) is a groundbreaking genomic technology that enables spatial localization analysis of gene expression within tissue sections. However, it is significantly limited by high costs and sparse spatial resolution. An alternative, more cost-effective strategy is to use deep learning methods to predict high-density gene expression profiles from histological images. However, existing methods struggle to capture rich image features effectively or rely on low-dimensional positional coordinates, making it difficult to accurately predict high-resolution gene expression profiles. To address these limitations, we developed HisToSGE, a method that employs a Pathology Image Large Model (PILM) to extract rich image features from histological images and utilizes a feature learning module to robustly generate high-resolution gene expression profiles. We evaluated HisToSGE on four ST datasets, comparing its performance with five state-of-the-art baseline methods. The results demonstrate that HisToSGE excels in generating high-resolution gene expression profiles and performing downstream tasks such as spatial domain identification. All code and public datasets used in this paper are available at https://github.com/wenwenmin/HisToSGE and https://zenodo.org/records/12792163.
stEnTrans: Transformer-based deep learning for spatial transcriptomics enhancement
Jul 11, 2024The spatial location of cells within tissues and organs is crucial for the manifestation of their specific functions.Spatial transcriptomics technology enables comprehensive measurement of the gene expression patterns in tissues while retaining spatial information. However, current popular spatial transcriptomics techniques either have shallow sequencing depth or low resolution. We present stEnTrans, a deep learning method based on Transformer architecture that provides comprehensive predictions for gene expression in unmeasured areas or unexpectedly lost areas and enhances gene expression in original and inputed spots. Utilizing a self-supervised learning approach, stEnTrans establishes proxy tasks on gene expression profile without requiring additional data, mining intrinsic features of the tissues as supervisory information. We evaluate stEnTrans on six datasets and the results indicate superior performance in enhancing spots resolution and predicting gene expression in unmeasured areas compared to other deep learning and traditional interpolation methods. Additionally, Our method also can help the discovery of spatial patterns in Spatial Transcriptomics and enrich to more biologically significant pathways. Our source code is available at https://github.com/shuailinxue/stEnTrans.