 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHigh-Resolution Spatial Transcriptomics from Histology Images using HisToSGE
Jul 30, 2024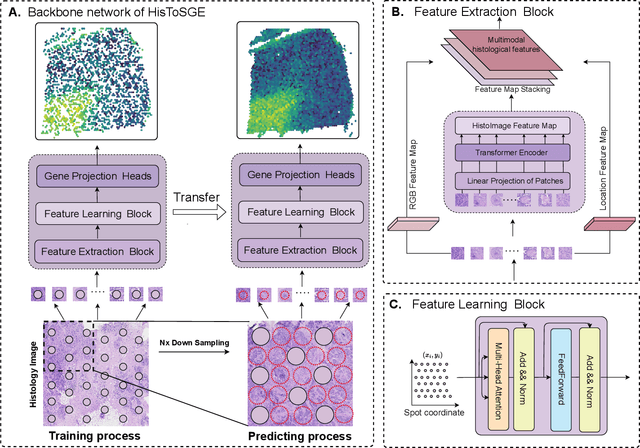
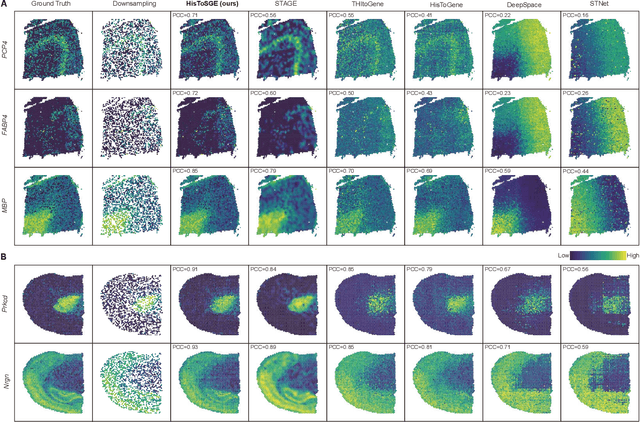
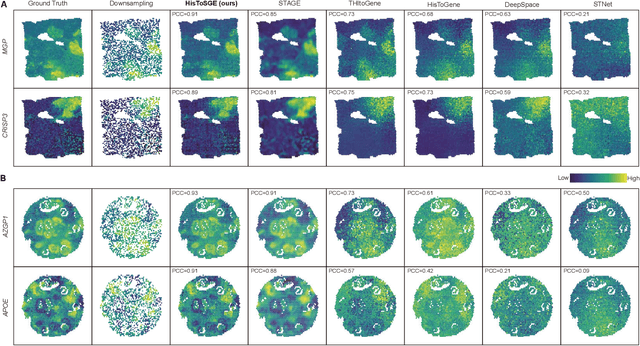
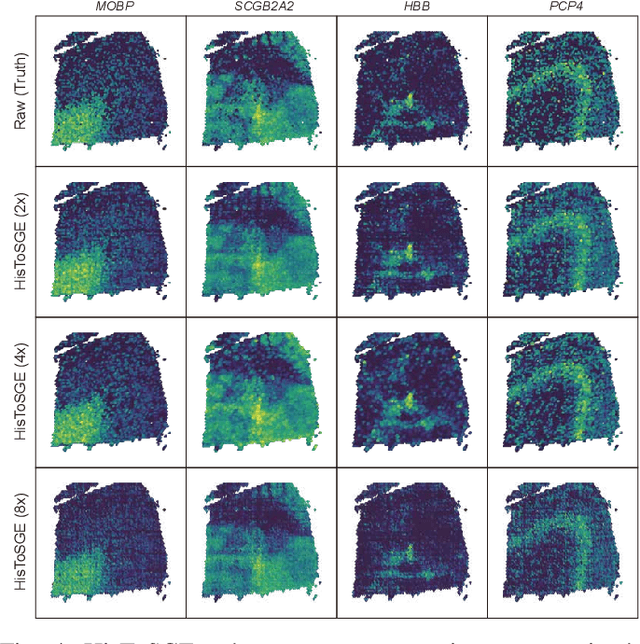
Spatial transcriptomics (ST) is a groundbreaking genomic technology that enables spatial localization analysis of gene expression within tissue sections. However, it is significantly limited by high costs and sparse spatial resolution. An alternative, more cost-effective strategy is to use deep learning methods to predict high-density gene expression profiles from histological images. However, existing methods struggle to capture rich image features effectively or rely on low-dimensional positional coordinates, making it difficult to accurately predict high-resolution gene expression profiles. To address these limitations, we developed HisToSGE, a method that employs a Pathology Image Large Model (PILM) to extract rich image features from histological images and utilizes a feature learning module to robustly generate high-resolution gene expression profiles. We evaluated HisToSGE on four ST datasets, comparing its performance with five state-of-the-art baseline methods. The results demonstrate that HisToSGE excels in generating high-resolution gene expression profiles and performing downstream tasks such as spatial domain identification. All code and public datasets used in this paper are available at https://github.com/wenwenmin/HisToSGE and https://zenodo.org/records/12792163.
Multimodal contrastive learning for spatial gene expression prediction using histology images
Jul 11, 2024



In recent years, the advent of spatial transcriptomics (ST) technology has unlocked unprecedented opportunities for delving into the complexities of gene expression patterns within intricate biological systems. Despite its transformative potential, the prohibitive cost of ST technology remains a significant barrier to its widespread adoption in large-scale studies. An alternative, more cost-effective strategy involves employing artificial intelligence to predict gene expression levels using readily accessible whole-slide images (WSIs) stained with Hematoxylin and Eosin (H\&E). However, existing methods have yet to fully capitalize on multimodal information provided by H&E images and ST data with spatial location. In this paper, we propose \textbf{mclSTExp}, a multimodal contrastive learning with Transformer and Densenet-121 encoder for Spatial Transcriptomics Expression prediction. We conceptualize each spot as a "word", integrating its intrinsic features with spatial context through the self-attention mechanism of a Transformer encoder. This integration is further enriched by incorporating image features via contrastive learning, thereby enhancing the predictive capability of our model. Our extensive evaluation of \textbf{mclSTExp} on two breast cancer datasets and a skin squamous cell carcinoma dataset demonstrates its superior performance in predicting spatial gene expression. Moreover, mclSTExp has shown promise in interpreting cancer-specific overexpressed genes, elucidating immune-related genes, and identifying specialized spatial domains annotated by pathologists. Our source code is available at https://github.com/shizhiceng/mclSTExp.