 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStyle-Aware Blending and Prototype-Based Cross-Contrast Consistency for Semi-Supervised Medical Image Segmentation
Jul 28, 2025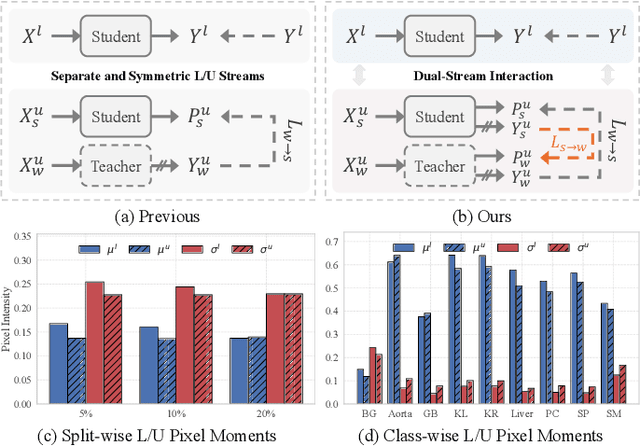



Weak-strong consistency learning strategies are widely employed in semi-supervised medical image segmentation to train models by leveraging limited labeled data and enforcing weak-to-strong consistency. However, existing methods primarily focus on designing and combining various perturbation schemes, overlooking the inherent potential and limitations within the framework itself. In this paper, we first identify two critical deficiencies: (1) separated training data streams, which lead to confirmation bias dominated by the labeled stream; and (2) incomplete utilization of supervisory information, which limits exploration of strong-to-weak consistency. To tackle these challenges, we propose a style-aware blending and prototype-based cross-contrast consistency learning framework. Specifically, inspired by the empirical observation that the distribution mismatch between labeled and unlabeled data can be characterized by statistical moments, we design a style-guided distribution blending module to break the independent training data streams. Meanwhile, considering the potential noise in strong pseudo-labels, we introduce a prototype-based cross-contrast strategy to encourage the model to learn informative supervisory signals from both weak-to-strong and strong-to-weak predictions, while mitigating the adverse effects of noise. Experimental results demonstrate the effectiveness and superiority of our framework across multiple medical segmentation benchmarks under various semi-supervised settings.
MSVM-UNet: Multi-Scale Vision Mamba UNet for Medical Image Segmentation
Aug 25, 2024



State Space Models (SSMs), especially Mamba, have shown great promise in medical image segmentation due to their ability to model long-range dependencies with linear computational complexity. However, accurate medical image segmentation requires the effective learning of both multi-scale detailed feature representations and global contextual dependencies. Although existing works have attempted to address this issue by integrating CNNs and SSMs to leverage their respective strengths, they have not designed specialized modules to effectively capture multi-scale feature representations, nor have they adequately addressed the directional sensitivity problem when applying Mamba to 2D image data. To overcome these limitations, we propose a Multi-Scale Vision Mamba UNet model for medical image segmentation, termed MSVM-UNet. Specifically, by introducing multi-scale convolutions in the VSS blocks, we can more effectively capture and aggregate multi-scale feature representations from the hierarchical features of the VMamba encoder and better handle 2D visual data. Additionally, the large kernel patch expanding (LKPE) layers achieve more efficient upsampling of feature maps by simultaneously integrating spatial and channel information. Extensive experiments on the Synapse and ACDC datasets demonstrate that our approach is more effective than some state-of-the-art methods in capturing and aggregating multi-scale feature representations and modeling long-range dependencies between pixels.
scASDC: Attention Enhanced Structural Deep Clustering for Single-cell RNA-seq Data
Aug 09, 2024



Single-cell RNA sequencing (scRNA-seq) data analysis is pivotal for understanding cellular heterogeneity. However, the high sparsity and complex noise patterns inherent in scRNA-seq data present significant challenges for traditional clustering methods. To address these issues, we propose a deep clustering method, Attention-Enhanced Structural Deep Embedding Graph Clustering (scASDC), which integrates multiple advanced modules to improve clustering accuracy and robustness.Our approach employs a multi-layer graph convolutional network (GCN) to capture high-order structural relationships between cells, termed as the graph autoencoder module. To mitigate the oversmoothing issue in GCNs, we introduce a ZINB-based autoencoder module that extracts content information from the data and learns latent representations of gene expression. These modules are further integrated through an attention fusion mechanism, ensuring effective combination of gene expression and structural information at each layer of the GCN. Additionally, a self-supervised learning module is incorporated to enhance the robustness of the learned embeddings. Extensive experiments demonstrate that scASDC outperforms existing state-of-the-art methods, providing a robust and effective solution for single-cell clustering tasks. Our method paves the way for more accurate and meaningful analysis of single-cell RNA sequencing data, contributing to better understanding of cellular heterogeneity and biological processes. All code and public datasets used in this paper are available at \url{https://github.com/wenwenmin/scASDC} and \url{https://zenodo.org/records/12814320}.
Masked adversarial neural network for cell type deconvolution in spatial transcriptomics
Aug 09, 2024



Accurately determining cell type composition in disease-relevant tissues is crucial for identifying disease targets. Most existing spatial transcriptomics (ST) technologies cannot achieve single-cell resolution, making it challenging to accurately determine cell types. To address this issue, various deconvolution methods have been developed. Most of these methods use single-cell RNA sequencing (scRNA-seq) data from the same tissue as a reference to infer cell types in ST data spots. However, they often overlook the differences between scRNA-seq and ST data. To overcome this limitation, we propose a Masked Adversarial Neural Network (MACD). MACD employs adversarial learning to align real ST data with simulated ST data generated from scRNA-seq data. By mapping them into a unified latent space, it can minimize the differences between the two types of data. Additionally, MACD uses masking techniques to effectively learn the features of real ST data and mitigate noise. We evaluated MACD on 32 simulated datasets and 2 real datasets, demonstrating its accuracy in performing cell type deconvolution. All code and public datasets used in this paper are available at https://github.com/wenwenmin/MACD and https://zenodo.org/records/12804822.
stMCDI: Masked Conditional Diffusion Model with Graph Neural Network for Spatial Transcriptomics Data Imputation
Mar 16, 2024



Spatially resolved transcriptomics represents a significant advancement in single-cell analysis by offering both gene expression data and their corresponding physical locations. However, this high degree of spatial resolution entails a drawback, as the resulting spatial transcriptomic data at the cellular level is notably plagued by a high incidence of missing values. Furthermore, most existing imputation methods either overlook the spatial information between spots or compromise the overall gene expression data distribution. To address these challenges, our primary focus is on effectively utilizing the spatial location information within spatial transcriptomic data to impute missing values, while preserving the overall data distribution. We introduce \textbf{stMCDI}, a novel conditional diffusion model for spatial transcriptomics data imputation, which employs a denoising network trained using randomly masked data portions as guidance, with the unmasked data serving as conditions. Additionally, it utilizes a GNN encoder to integrate the spatial position information, thereby enhancing model performance. The results obtained from spatial transcriptomics datasets elucidate the performance of our methods relative to existing approaches.