 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeComparative evaluation of photogrammetric reconstruction methods and 3D Gaussian Splatting for road surface roughness analysis
May 28, 2026Image-based 3D reconstruction offers a low-cost alternative to traditional sensor-based techniques for road surface assessment. This study compares four reconstruction pipelines--COLMAP, Meshroom, Metashape, and 3D Gaussian Splatting (3DGS)--to evaluate their ability to estimate road surface roughness from smartphone imagery. All point clouds were processed in CloudCompare using a consistent workflow involving orientation alignment, segmentation, normal estimation, and roughness computation at neighborhood radiuses of 0.2, 0.4, and 0.6 model units. The results show that COLMAP provides the highest sensitivity to micro-texture, while Meshroom yields balanced reconstructions with moderate roughness variation. Metashape produces the smoothest geometry due to its internal filtering, and 3DGS captures visible irregularities but exhibits higher noise and lower density. The comparison demonstrates that open-source pipelines are viable for relative roughness evaluation, offering a practical approach for low-cost pavement monitoring.
EvoLM: Self-Evolving Language Models through Co-Evolved Discriminative Rubrics
May 05, 2026Language models encode substantial evaluative knowledge from pretraining, yet current post-training methods rely on external supervision (human annotations, proprietary models, or scalar reward models) to produce reward signals. Each imposes a ceiling. Human judgment cannot supervise capabilities beyond its own, proprietary APIs create dependencies, and verifiable rewards cover only domains with ground-truth answers. Self-improvement from a model's own evaluative capacity is a reward source that scales with the model itself, yet remains largely untapped by current methods. We introduce EVOLM, a post-training method that structures this capacity into explicit discriminative rubrics and uses them as training signal. EVOLM trains two capabilities within a single language model in alternation: (1) a rubric generator producing instance-specific evaluation criteria optimized for discriminative utility, which maximizes a small frozen judge's ability to distinguish preferred from dispreferred responses; and (2) a policy trained using those rubric-conditioned scores as reward. All preference signals are constructed from the policy's own outputs via temporal contrast with earlier checkpoints, requiring no human annotation or external supervision. EVOLM trains a Qwen3-8B model to generate rubrics that outperform GPT-4.1 on RewardBench-2 by 25.7%. The co-trained policy achieves 69.3% average on the OLMo3-Adapt suite, outperforming policies trained with GPT-4.1 prompted rubrics by 3.9% and with the state-of-the-art 8B reward model SkyWork-RM by 16%. Overall, EVOLM demonstrates that structuring a model's evaluative capacity into co-evolving discriminative rubrics enables self-improvement without external supervision.
Meta-Reinforcement Learning with Self-Reflection for Agentic Search
Mar 11, 2026This paper introduces MR-Search, an in-context meta reinforcement learning (RL) formulation for agentic search with self-reflection. Instead of optimizing a policy within a single independent episode with sparse rewards, MR-Search trains a policy that conditions on past episodes and adapts its search strategy across episodes. MR-Search learns to learn a search strategy with self-reflection, allowing search agents to improve in-context exploration at test-time. Specifically, MR-Search performs cross-episode exploration by generating explicit self-reflections after each episode and leveraging them as additional context to guide subsequent attempts, thereby promoting more effective exploration during test-time. We further introduce a multi-turn RL algorithm that estimates a dense relative advantage at the turn level, enabling fine-grained credit assignment on each episode. Empirical results across various benchmarks demonstrate the advantages of MR-Search over baselines based RL, showing strong generalization and relative improvements of 9.2% to 19.3% across eight benchmarks. Our code and data are available at https://github.com/tengxiao1/MR-Search.
Small Reward Models via Backward Inference
Feb 14, 2026Reward models (RMs) play a central role throughout the language model (LM) pipeline, particularly in non-verifiable domains. However, the dominant LLM-as-a-Judge paradigm relies on the strong reasoning capabilities of large models, while alternative approaches require reference responses or explicit rubrics, limiting flexibility and broader accessibility. In this work, we propose FLIP (FLipped Inference for Prompt reconstruction), a reference-free and rubric-free reward modeling approach that reformulates reward modeling through backward inference: inferring the instruction that would most plausibly produce a given response. The similarity between the inferred and the original instructions is then used as the reward signal. Evaluations across four domains using 13 small language models show that FLIP outperforms LLM-as-a-Judge baselines by an average of 79.6%. Moreover, FLIP substantially improves downstream performance in extrinsic evaluations under test-time scaling via parallel sampling and GRPO training. We further find that FLIP is particularly effective for longer outputs and robust to common forms of reward hacking. By explicitly exploiting the validation-generation gap, FLIP enables reliable reward modeling in downscaled regimes where judgment methods fail. Code available at https://github.com/yikee/FLIP.
Olmo 3
Dec 15, 2025We introduce Olmo 3, a family of state-of-the-art, fully-open language models at the 7B and 32B parameter scales. Olmo 3 model construction targets long-context reasoning, function calling, coding, instruction following, general chat, and knowledge recall. This release includes the entire model flow, i.e., the full lifecycle of the family of models, including every stage, checkpoint, data point, and dependency used to build it. Our flagship model, Olmo 3 Think 32B, is the strongest fully-open thinking model released to-date.
Simple Denoising Diffusion Language Models
Oct 27, 2025Diffusion models have recently been extended to language generation through Masked Diffusion Language Models (MDLMs), which achieve performance competitive with strong autoregressive models. However, MDLMs tend to degrade in the few-step regime and cannot directly adopt existing few-step distillation methods designed for continuous diffusion models, as they lack the intrinsic property of mapping from noise to data. Recent Uniform-state Diffusion Models (USDMs), initialized from a uniform prior, alleviate some limitations but still suffer from complex loss formulations that hinder scalability. In this work, we propose a simplified denoising-based loss for USDMs that optimizes only noise-replaced tokens, stabilizing training and matching ELBO-level performance. Furthermore, by framing denoising as self-supervised learning, we introduce a simple modification to our denoising loss with contrastive-inspired negative gradients, which is practical and yield additional improvements in generation quality.
Incentivizing Strong Reasoning from Weak Supervision
May 28, 2025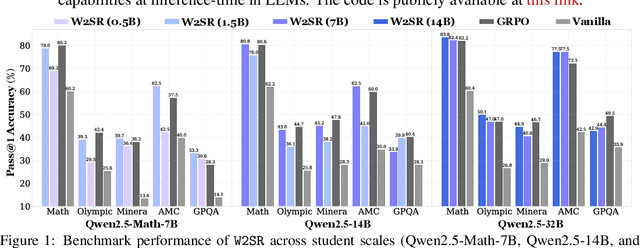
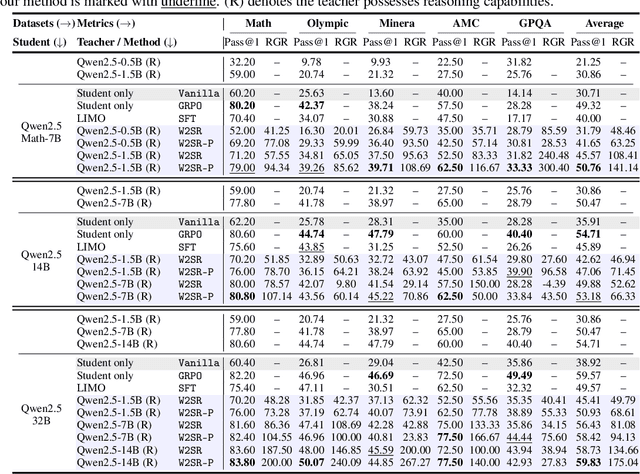
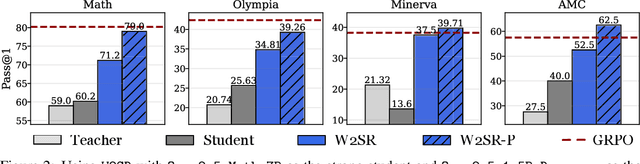
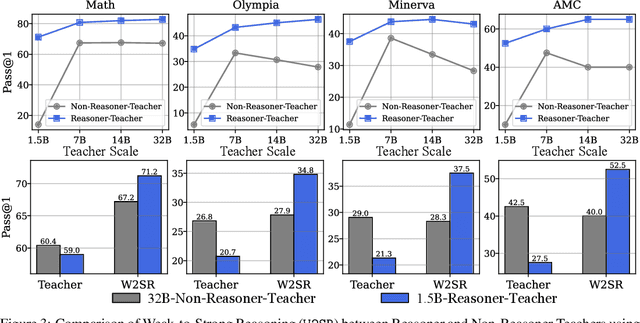
Large language models (LLMs) have demonstrated impressive performance on reasoning-intensive tasks, but enhancing their reasoning abilities typically relies on either reinforcement learning (RL) with verifiable signals or supervised fine-tuning (SFT) with high-quality long chain-of-thought (CoT) demonstrations, both of which are expensive. In this paper, we study a novel problem of incentivizing the reasoning capacity of LLMs without expensive high-quality demonstrations and reinforcement learning. We investigate whether the reasoning capabilities of LLMs can be effectively incentivized via supervision from significantly weaker models. We further analyze when and why such weak supervision succeeds in eliciting reasoning abilities in stronger models. Our findings show that supervision from significantly weaker reasoners can substantially improve student reasoning performance, recovering close to 94% of the gains of expensive RL at a fraction of the cost. Experiments across diverse benchmarks and model architectures demonstrate that weak reasoners can effectively incentivize reasoning in stronger student models, consistently improving performance across a wide range of reasoning tasks. Our results suggest that this simple weak-to-strong paradigm is a promising and generalizable alternative to costly methods for incentivizing strong reasoning capabilities at inference-time in LLMs. The code is publicly available at https://github.com/yuanyige/w2sr.
Inference-time Alignment in Continuous Space
May 26, 2025Aligning large language models with human feedback at inference time has received increasing attention due to its flexibility. Existing methods rely on generating multiple responses from the base policy for search using a reward model, which can be considered as searching in a discrete response space. However, these methods struggle to explore informative candidates when the base policy is weak or the candidate set is small, resulting in limited effectiveness. In this paper, to address this problem, we propose Simple Energy Adaptation ($\textbf{SEA}$), a simple yet effective algorithm for inference-time alignment. In contrast to expensive search over the discrete space, SEA directly adapts original responses from the base policy toward the optimal one via gradient-based sampling in continuous latent space. Specifically, SEA formulates inference as an iterative optimization procedure on an energy function over actions in the continuous space defined by the optimal policy, enabling simple and effective alignment. For instance, despite its simplicity, SEA outperforms the second-best baseline with a relative improvement of up to $ \textbf{77.51%}$ on AdvBench and $\textbf{16.36%}$ on MATH. Our code is publicly available at https://github.com/yuanyige/SEA
Incentivizing Reasoning from Weak Supervision
May 26, 2025Large language models (LLMs) have demonstrated impressive performance on reasoning-intensive tasks, but enhancing their reasoning abilities typically relies on either reinforcement learning (RL) with verifiable signals or supervised fine-tuning (SFT) with high-quality long chain-of-thought (CoT) demonstrations, both of which are expensive. In this paper, we study a novel problem of incentivizing the reasoning capacity of LLMs without expensive high-quality demonstrations and reinforcement learning. We investigate whether the reasoning capabilities of LLMs can be effectively incentivized via supervision from significantly weaker models. We further analyze when and why such weak supervision succeeds in eliciting reasoning abilities in stronger models. Our findings show that supervision from significantly weaker reasoners can substantially improve student reasoning performance, recovering close to 94% of the gains of expensive RL at a fraction of the cost. Experiments across diverse benchmarks and model architectures demonstrate that weak reasoners can effectively incentivize reasoning in stronger student models, consistently improving performance across a wide range of reasoning tasks. Our results suggest that this simple weak-to-strong paradigm is a promising and generalizable alternative to costly methods for incentivizing strong reasoning capabilities at inference-time in LLMs. The code is publicly available at https://github.com/yuanyige/W2SR.
InfoPO: On Mutual Information Maximization for Large Language Model Alignment
May 13, 2025We study the post-training of large language models (LLMs) with human preference data. Recently, direct preference optimization and its variants have shown considerable promise in aligning language models, eliminating the need for reward models and online sampling. Despite these benefits, these methods rely on explicit assumptions about the Bradley-Terry (BT) model, which makes them prone to overfitting and results in suboptimal performance, particularly on reasoning-heavy tasks. To address these challenges, we propose a principled preference fine-tuning algorithm called InfoPO, which effectively and efficiently aligns large language models using preference data. InfoPO eliminates the reliance on the BT model and prevents the likelihood of the chosen response from decreasing. Extensive experiments confirm that InfoPO consistently outperforms established baselines on widely used open benchmarks, particularly in reasoning tasks.