 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMICCAI-CDMRI 2023 QuantConn Challenge Findings on Achieving Robust Quantitative Connectivity through Harmonized Preprocessing of Diffusion MRI
Nov 14, 2024



White matter alterations are increasingly implicated in neurological diseases and their progression. International-scale studies use diffusion-weighted magnetic resonance imaging (DW-MRI) to qualitatively identify changes in white matter microstructure and connectivity. Yet, quantitative analysis of DW-MRI data is hindered by inconsistencies stemming from varying acquisition protocols. There is a pressing need to harmonize the preprocessing of DW-MRI datasets to ensure the derivation of robust quantitative diffusion metrics across acquisitions. In the MICCAI-CDMRI 2023 QuantConn challenge, participants were provided raw data from the same individuals collected on the same scanner but with two different acquisitions and tasked with preprocessing the DW-MRI to minimize acquisition differences while retaining biological variation. Submissions are evaluated on the reproducibility and comparability of cross-acquisition bundle-wise microstructure measures, bundle shape features, and connectomics. The key innovations of the QuantConn challenge are that (1) we assess bundles and tractography in the context of harmonization for the first time, (2) we assess connectomics in the context of harmonization for the first time, and (3) we have 10x additional subjects over prior harmonization challenge, MUSHAC and 100x over SuperMUDI. We find that bundle surface area, fractional anisotropy, connectome assortativity, betweenness centrality, edge count, modularity, nodal strength, and participation coefficient measures are most biased by acquisition and that machine learning voxel-wise correction, RISH mapping, and NeSH methods effectively reduce these biases. In addition, microstructure measures AD, MD, RD, bundle length, connectome density, efficiency, and path length are least biased by these acquisition differences.
* Accepted for publication at the Journal of Machine Learning for Biomedical Imaging (MELBA) https://melba-journal.org/2024/019
DenseNet and Support Vector Machine classifications of major depressive disorder using vertex-wise cortical features
Nov 18, 2023



Major depressive disorder (MDD) is a complex psychiatric disorder that affects the lives of hundreds of millions of individuals around the globe. Even today, researchers debate if morphological alterations in the brain are linked to MDD, likely due to the heterogeneity of this disorder. The application of deep learning tools to neuroimaging data, capable of capturing complex non-linear patterns, has the potential to provide diagnostic and predictive biomarkers for MDD. However, previous attempts to demarcate MDD patients and healthy controls (HC) based on segmented cortical features via linear machine learning approaches have reported low accuracies. In this study, we used globally representative data from the ENIGMA-MDD working group containing an extensive sample of people with MDD (N=2,772) and HC (N=4,240), which allows a comprehensive analysis with generalizable results. Based on the hypothesis that integration of vertex-wise cortical features can improve classification performance, we evaluated the classification of a DenseNet and a Support Vector Machine (SVM), with the expectation that the former would outperform the latter. As we analyzed a multi-site sample, we additionally applied the ComBat harmonization tool to remove potential nuisance effects of site. We found that both classifiers exhibited close to chance performance (balanced accuracy DenseNet: 51%; SVM: 53%), when estimated on unseen sites. Slightly higher classification performance (balanced accuracy DenseNet: 58%; SVM: 55%) was found when the cross-validation folds contained subjects from all sites, indicating site effect. In conclusion, the integration of vertex-wise morphometric features and the use of the non-linear classifier did not lead to the differentiability between MDD and HC. Our results support the notion that MDD classification on this combination of features and classifiers is unfeasible.
Tackling the dimensions in imaging genetics with CLUB-PLS
Sep 20, 2023A major challenge in imaging genetics and similar fields is to link high-dimensional data in one domain, e.g., genetic data, to high dimensional data in a second domain, e.g., brain imaging data. The standard approach in the area are mass univariate analyses across genetic factors and imaging phenotypes. That entails executing one genome-wide association study (GWAS) for each pre-defined imaging measure. Although this approach has been tremendously successful, one shortcoming is that phenotypes must be pre-defined. Consequently, effects that are not confined to pre-selected regions of interest or that reflect larger brain-wide patterns can easily be missed. In this work we introduce a Partial Least Squares (PLS)-based framework, which we term Cluster-Bootstrap PLS (CLUB-PLS), that can work with large input dimensions in both domains as well as with large sample sizes. One key factor of the framework is to use cluster bootstrap to provide robust statistics for single input features in both domains. We applied CLUB-PLS to investigating the genetic basis of surface area and cortical thickness in a sample of 33,000 subjects from the UK Biobank. We found 107 genome-wide significant locus-phenotype pairs that are linked to 386 different genes. We found that a vast majority of these loci could be technically validated at a high rate: using classic GWAS or Genome-Wide Inferred Statistics (GWIS) we found that 85 locus-phenotype pairs exceeded the genome-wide suggestive (P<1e-05) threshold.
Linking Symptom Inventories using Semantic Textual Similarity
Sep 08, 2023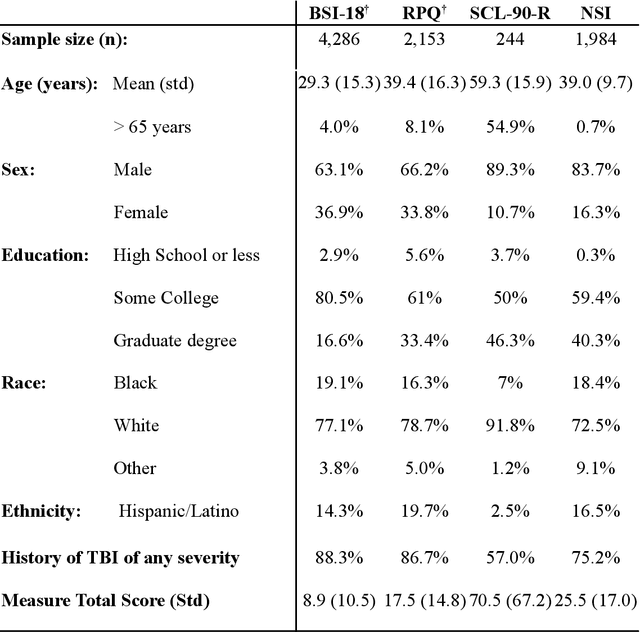
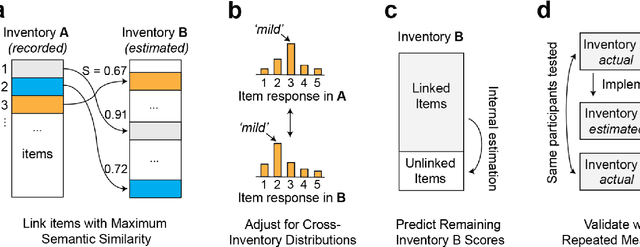
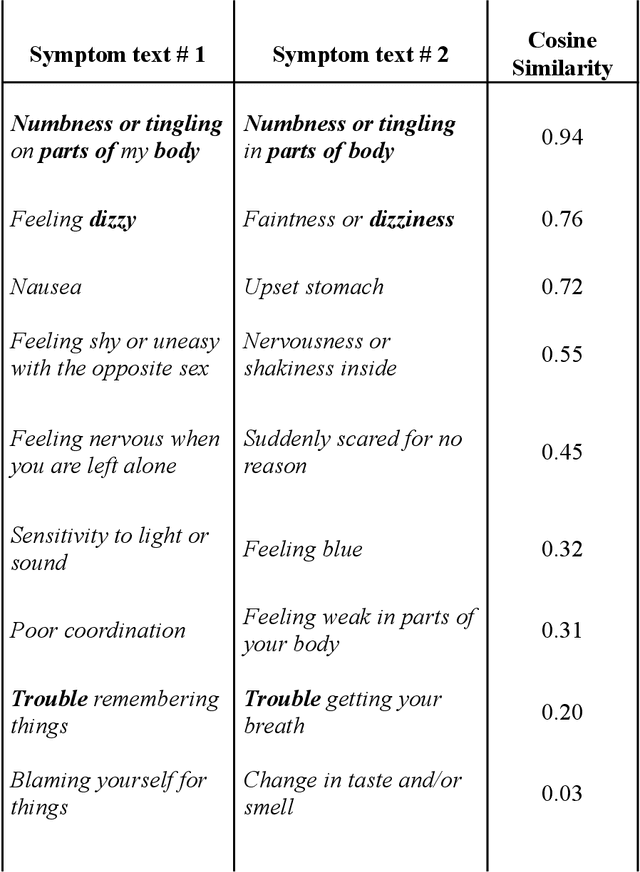
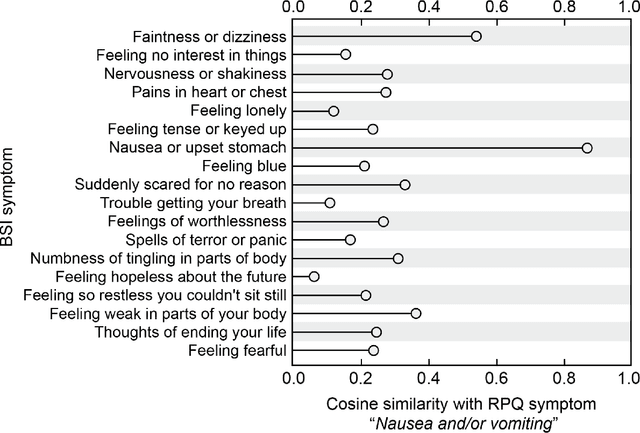
An extensive library of symptom inventories has been developed over time to measure clinical symptoms, but this variety has led to several long standing issues. Most notably, results drawn from different settings and studies are not comparable, which limits reproducibility. Here, we present an artificial intelligence (AI) approach using semantic textual similarity (STS) to link symptoms and scores across previously incongruous symptom inventories. We tested the ability of four pre-trained STS models to screen thousands of symptom description pairs for related content - a challenging task typically requiring expert panels. Models were tasked to predict symptom severity across four different inventories for 6,607 participants drawn from 16 international data sources. The STS approach achieved 74.8% accuracy across five tasks, outperforming other models tested. This work suggests that incorporating contextual, semantic information can assist expert decision-making processes, yielding gains for both general and disease-specific clinical assessment.
Simultaneous Matrix Diagonalization for Structural Brain Networks Classification
Oct 14, 2017
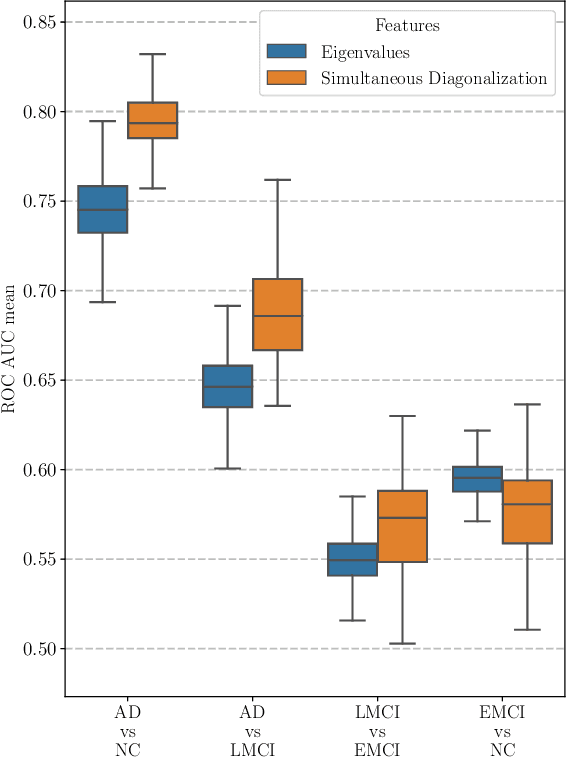

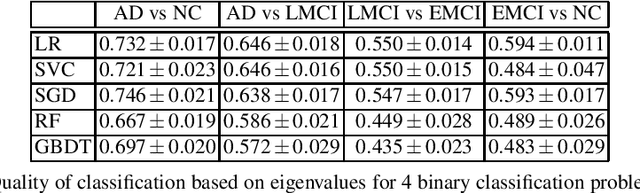
This paper considers the problem of brain disease classification based on connectome data. A connectome is a network representation of a human brain. The typical connectome classification problem is very challenging because of the small sample size and high dimensionality of the data. We propose to use simultaneous approximate diagonalization of adjacency matrices in order to compute their eigenstructures in more stable way. The obtained approximate eigenvalues are further used as features for classification. The proposed approach is demonstrated to be efficient for detection of Alzheimer's disease, outperforming simple baselines and competing with state-of-the-art approaches to brain disease classification.
Evaluating 35 Methods to Generate Structural Connectomes Using Pairwise Classification
Jun 19, 2017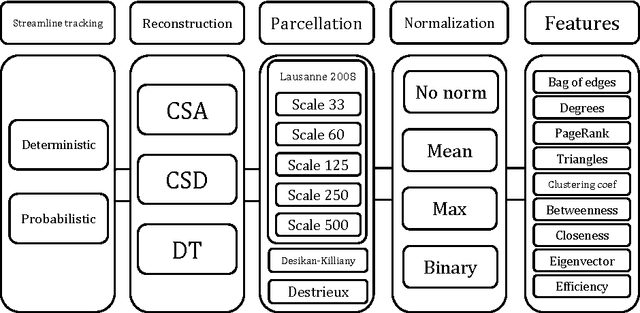
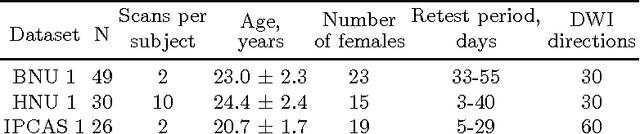

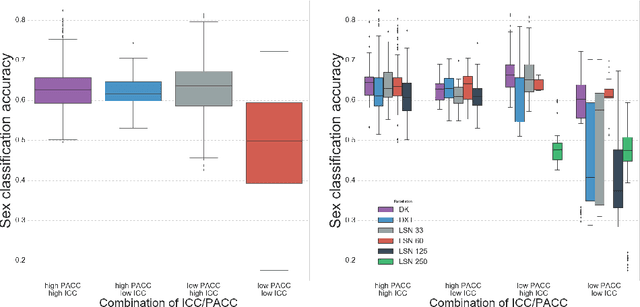
There is no consensus on how to construct structural brain networks from diffusion MRI. How variations in pre-processing steps affect network reliability and its ability to distinguish subjects remains opaque. In this work, we address this issue by comparing 35 structural connectome-building pipelines. We vary diffusion reconstruction models, tractography algorithms and parcellations. Next, we classify structural connectome pairs as either belonging to the same individual or not. Connectome weights and eight topological derivative measures form our feature set. For experiments, we use three test-retest datasets from the Consortium for Reliability and Reproducibility (CoRR) comprised of a total of 105 individuals. We also compare pairwise classification results to a commonly used parametric test-retest measure, Intraclass Correlation Coefficient (ICC).
Classification of Major Depressive Disorder via Multi-Site Weighted LASSO Model
Jun 03, 2017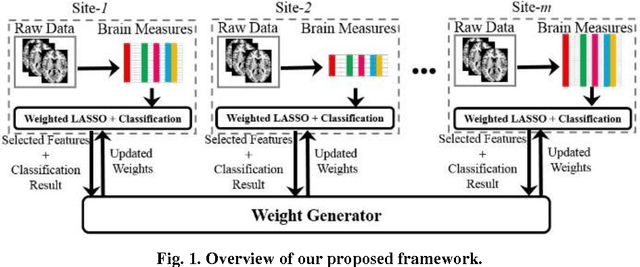
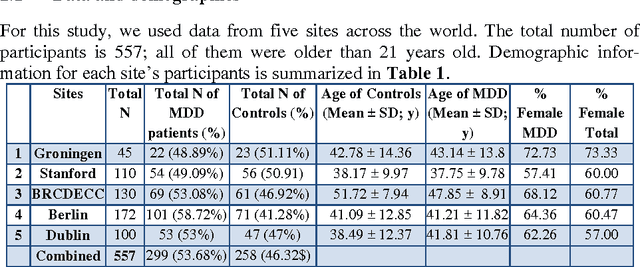

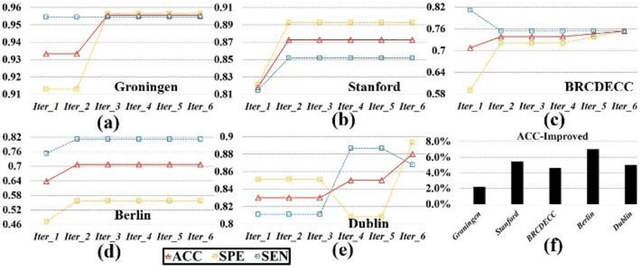
Large-scale collaborative analysis of brain imaging data, in psychiatry and neu-rology, offers a new source of statistical power to discover features that boost ac-curacy in disease classification, differential diagnosis, and outcome prediction. However, due to data privacy regulations or limited accessibility to large datasets across the world, it is challenging to efficiently integrate distributed information. Here we propose a novel classification framework through multi-site weighted LASSO: each site performs an iterative weighted LASSO for feature selection separately. Within each iteration, the classification result and the selected features are collected to update the weighting parameters for each feature. This new weight is used to guide the LASSO process at the next iteration. Only the fea-tures that help to improve the classification accuracy are preserved. In tests on da-ta from five sites (299 patients with major depressive disorder (MDD) and 258 normal controls), our method boosted classification accuracy for MDD by 4.9% on average. This result shows the potential of the proposed new strategy as an ef-fective and practical collaborative platform for machine learning on large scale distributed imaging and biobank data.
Large-scale Feature Selection of Risk Genetic Factors for Alzheimer's Disease via Distributed Group Lasso Regression
Apr 27, 2017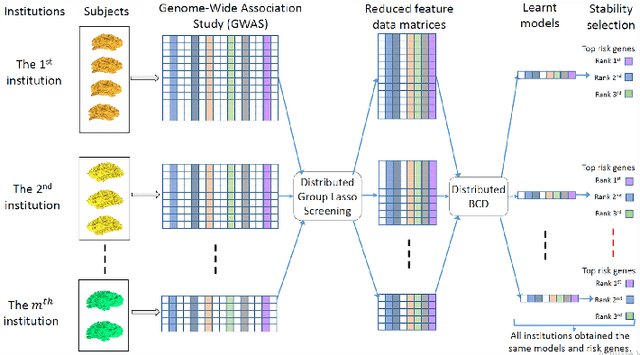
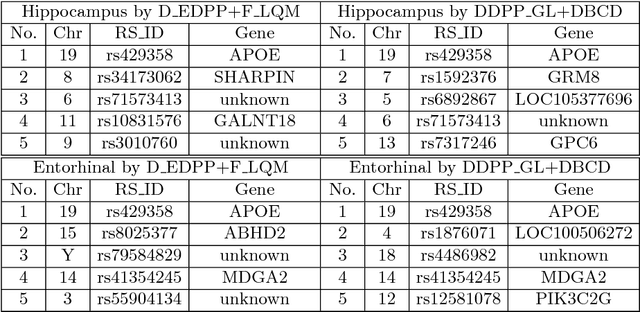
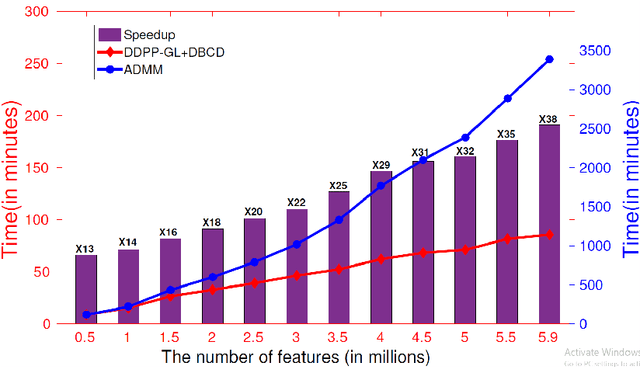
Genome-wide association studies (GWAS) have achieved great success in the genetic study of Alzheimer's disease (AD). Collaborative imaging genetics studies across different research institutions show the effectiveness of detecting genetic risk factors. However, the high dimensionality of GWAS data poses significant challenges in detecting risk SNPs for AD. Selecting relevant features is crucial in predicting the response variable. In this study, we propose a novel Distributed Feature Selection Framework (DFSF) to conduct the large-scale imaging genetics studies across multiple institutions. To speed up the learning process, we propose a family of distributed group Lasso screening rules to identify irrelevant features and remove them from the optimization. Then we select the relevant group features by performing the group Lasso feature selection process in a sequence of parameters. Finally, we employ the stability selection to rank the top risk SNPs that might help detect the early stage of AD. To the best of our knowledge, this is the first distributed feature selection model integrated with group Lasso feature selection as well as detecting the risk genetic factors across multiple research institutions system. Empirical studies are conducted on 809 subjects with 5.9 million SNPs which are distributed across several individual institutions, demonstrating the efficiency and effectiveness of the proposed method.
A Restaurant Process Mixture Model for Connectivity Based Parcellation of the Cortex
Mar 02, 2017
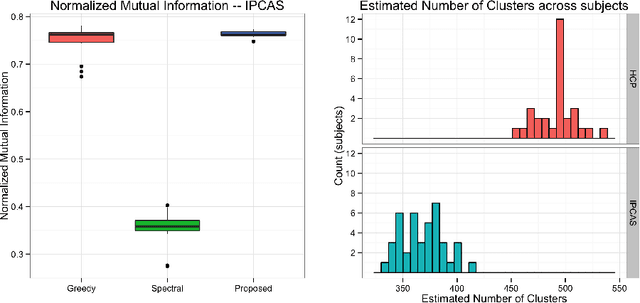
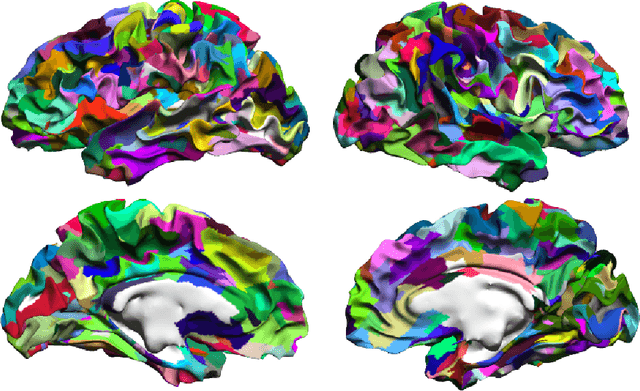
One of the primary objectives of human brain mapping is the division of the cortical surface into functionally distinct regions, i.e. parcellation. While it is generally agreed that at macro-scale different regions of the cortex have different functions, the exact number and configuration of these regions is not known. Methods for the discovery of these regions are thus important, particularly as the volume of available information grows. Towards this end, we present a parcellation method based on a Bayesian non-parametric mixture model of cortical connectivity.
Structural Connectome Validation Using Pairwise Classification
Jan 30, 2017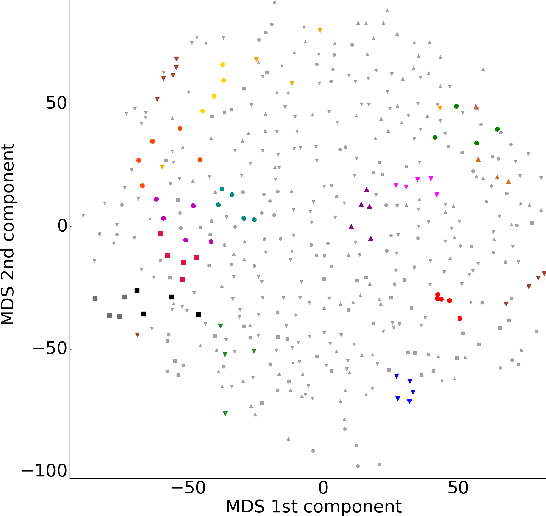
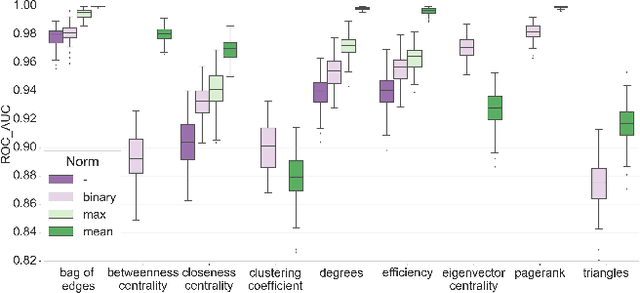
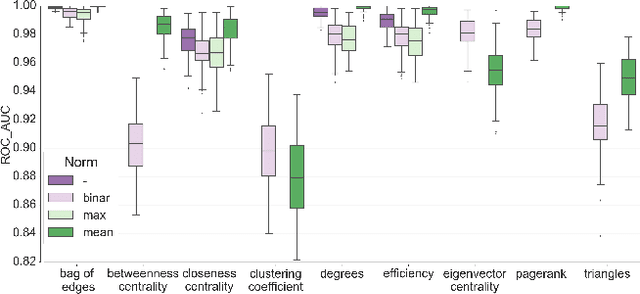
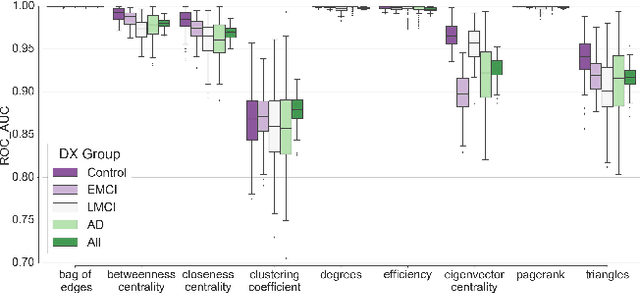
In this work, we study the extent to which structural connectomes and topological derivative measures are unique to individual changes within human brains. To do so, we classify structural connectome pairs from two large longitudinal datasets as either belonging to the same individual or not. Our data is comprised of 227 individuals from the Alzheimer's Disease Neuroimaging Initiative (ADNI) and 226 from the Parkinson's Progression Markers Initiative (PPMI). We achieve 0.99 area under the ROC curve score for features which represent either weights or network structure of the connectomes (node degrees, PageRank and local efficiency). Our approach may be useful for eliminating noisy features as a preprocessing step in brain aging studies and early diagnosis classification problems.