 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA computationally frugal open-source foundation model for thoracic disease detection in lung cancer screening programs
Jul 02, 2025Low-dose computed tomography (LDCT) imaging employed in lung cancer screening (LCS) programs is increasing in uptake worldwide. LCS programs herald a generational opportunity to simultaneously detect cancer and non-cancer-related early-stage lung disease. Yet these efforts are hampered by a shortage of radiologists to interpret scans at scale. Here, we present TANGERINE, a computationally frugal, open-source vision foundation model for volumetric LDCT analysis. Designed for broad accessibility and rapid adaptation, TANGERINE can be fine-tuned off the shelf for a wide range of disease-specific tasks with limited computational resources and training data. Relative to models trained from scratch, TANGERINE demonstrates fast convergence during fine-tuning, thereby requiring significantly fewer GPU hours, and displays strong label efficiency, achieving comparable or superior performance with a fraction of fine-tuning data. Pretrained using self-supervised learning on over 98,000 thoracic LDCTs, including the UK's largest LCS initiative to date and 27 public datasets, TANGERINE achieves state-of-the-art performance across 14 disease classification tasks, including lung cancer and multiple respiratory diseases, while generalising robustly across diverse clinical centres. By extending a masked autoencoder framework to 3D imaging, TANGERINE offers a scalable solution for LDCT analysis, departing from recent closed, resource-intensive models by combining architectural simplicity, public availability, and modest computational requirements. Its accessible, open-source lightweight design lays the foundation for rapid integration into next-generation medical imaging tools that could transform LCS initiatives, allowing them to pivot from a singular focus on lung cancer detection to comprehensive respiratory disease management in high-risk populations.
Disentangled Diffusion Autoencoder for Harmonization of Multi-site Neuroimaging Data
Aug 28, 2024



Combining neuroimaging datasets from multiple sites and scanners can help increase statistical power and thus provide greater insight into subtle neuroanatomical effects. However, site-specific effects pose a challenge by potentially obscuring the biological signal and introducing unwanted variance. Existing harmonization techniques, which use statistical models to remove such effects, have been shown to incompletely remove site effects while also failing to preserve biological variability. More recently, generative models using GANs or autoencoder-based approaches, have been proposed for site adjustment. However, such methods are known for instability during training or blurry image generation. In recent years, diffusion models have become increasingly popular for their ability to generate high-quality synthetic images. In this work, we introduce the disentangled diffusion autoencoder (DDAE), a novel diffusion model designed for controlling specific aspects of an image. We apply the DDAE to the task of harmonizing MR images by generating high-quality site-adjusted images that preserve biological variability. We use data from 7 different sites and demonstrate the DDAE's superiority in generating high-resolution, harmonized 2D MR images over previous approaches. As far as we are aware, this work marks the first diffusion-based model for site adjustment of neuroimaging data.
A tutorial on multi-view autoencoders using the multi-view-AE library
Mar 12, 2024


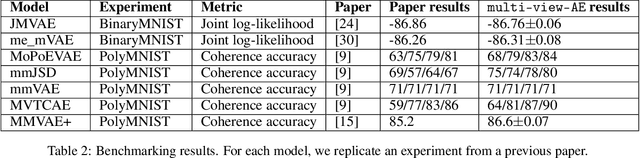
There has been a growing interest in recent years in modelling multiple modalities (or views) of data to for example, understand the relationship between modalities or to generate missing data. Multi-view autoencoders have gained significant traction for their adaptability and versatility in modelling multi-modal data, demonstrating an ability to tailor their approach to suit the characteristics of the data at hand. However, most multi-view autoencoders have inconsistent notation and are often implemented using different coding frameworks. To address this, we present a unified mathematical framework for multi-view autoencoders, consolidating their formulations. Moreover, we offer insights into the motivation and theoretical advantages of each model. To facilitate accessibility and practical use, we extend the documentation and functionality of the previously introduced \texttt{multi-view-AE} library. This library offers Python implementations of numerous multi-view autoencoder models, presented within a user-friendly framework. Through benchmarking experiments, we evaluate our implementations against previous ones, demonstrating comparable or superior performance. This work aims to establish a cohesive foundation for multi-modal modelling, serving as a valuable educational resource in the field.
Tackling the dimensions in imaging genetics with CLUB-PLS
Sep 20, 2023A major challenge in imaging genetics and similar fields is to link high-dimensional data in one domain, e.g., genetic data, to high dimensional data in a second domain, e.g., brain imaging data. The standard approach in the area are mass univariate analyses across genetic factors and imaging phenotypes. That entails executing one genome-wide association study (GWAS) for each pre-defined imaging measure. Although this approach has been tremendously successful, one shortcoming is that phenotypes must be pre-defined. Consequently, effects that are not confined to pre-selected regions of interest or that reflect larger brain-wide patterns can easily be missed. In this work we introduce a Partial Least Squares (PLS)-based framework, which we term Cluster-Bootstrap PLS (CLUB-PLS), that can work with large input dimensions in both domains as well as with large sample sizes. One key factor of the framework is to use cluster bootstrap to provide robust statistics for single input features in both domains. We applied CLUB-PLS to investigating the genetic basis of surface area and cortical thickness in a sample of 33,000 subjects from the UK Biobank. We found 107 genome-wide significant locus-phenotype pairs that are linked to 386 different genes. We found that a vast majority of these loci could be technically validated at a high rate: using classic GWAS or Genome-Wide Inferred Statistics (GWIS) we found that 85 locus-phenotype pairs exceeded the genome-wide suggestive (P<1e-05) threshold.
Multi-modal Variational Autoencoders for normative modelling across multiple imaging modalities
Mar 16, 2023


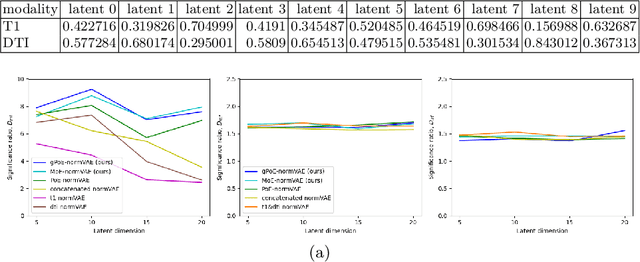
One of the challenges of studying common neurological disorders is disease heterogeneity including differences in causes, neuroimaging characteristics, comorbidities, or genetic variation. Normative modelling has become a popular method for studying such cohorts where the 'normal' behaviour of a physiological system is modelled and can be used at subject level to detect deviations relating to disease pathology. For many heterogeneous diseases, we expect to observe abnormalities across a range of neuroimaging and biological variables. However, thus far, normative models have largely been developed for studying a single imaging modality. We aim to develop a multi-modal normative modelling framework where abnormality is aggregated across variables of multiple modalities and is better able to detect deviations than uni-modal baselines. We propose two multi-modal VAE normative models to detect subject level deviations across T1 and DTI data. Our proposed models were better able to detect diseased individuals, capture disease severity, and correlate with patient cognition than baseline approaches. We also propose a multivariate latent deviation metric, measuring deviations from the joint latent space, which outperformed feature-based metrics.
Development and evaluation of intraoperative ultrasound segmentation with negative image frames and multiple observer labels
Jul 28, 2021
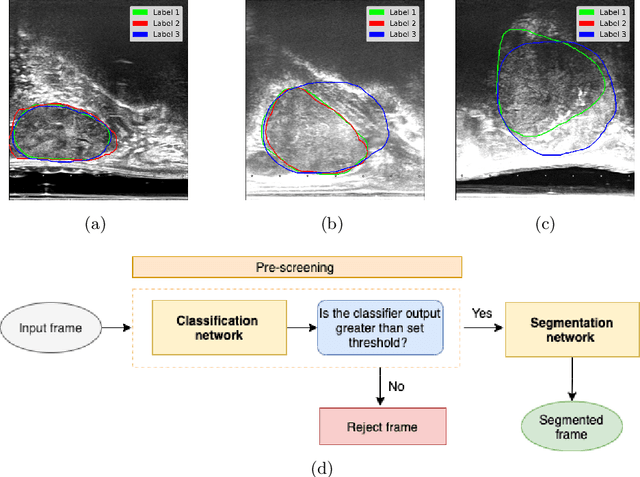
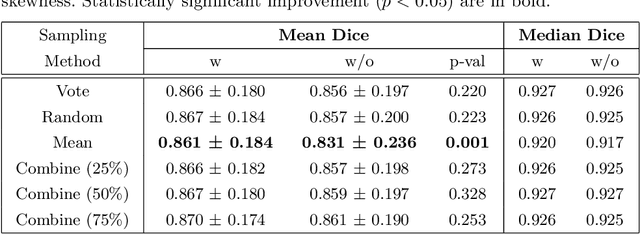
When developing deep neural networks for segmenting intraoperative ultrasound images, several practical issues are encountered frequently, such as the presence of ultrasound frames that do not contain regions of interest and the high variance in ground-truth labels. In this study, we evaluate the utility of a pre-screening classification network prior to the segmentation network. Experimental results demonstrate that such a classifier, minimising frame classification errors, was able to directly impact the number of false positive and false negative frames. Importantly, the segmentation accuracy on the classifier-selected frames, that would be segmented, remains comparable to or better than those from standalone segmentation networks. Interestingly, the efficacy of the pre-screening classifier was affected by the sampling methods for training labels from multiple observers, a seemingly independent problem. We show experimentally that a previously proposed approach, combining random sampling and consensus labels, may need to be adapted to perform well in our application. Furthermore, this work aims to share practical experience in developing a machine learning application that assists highly variable interventional imaging for prostate cancer patients, to present robust and reproducible open-source implementations, and to report a set of comprehensive results and analysis comparing these practical, yet important, options in a real-world clinical application.
Federated Learning in Distributed Medical Databases: Meta-Analysis of Large-Scale Subcortical Brain Data
Oct 22, 2018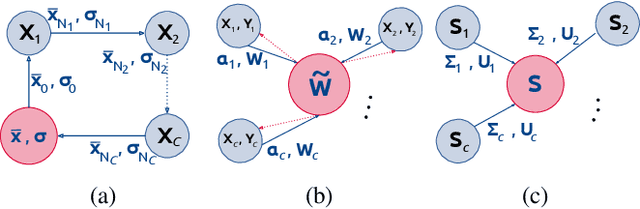

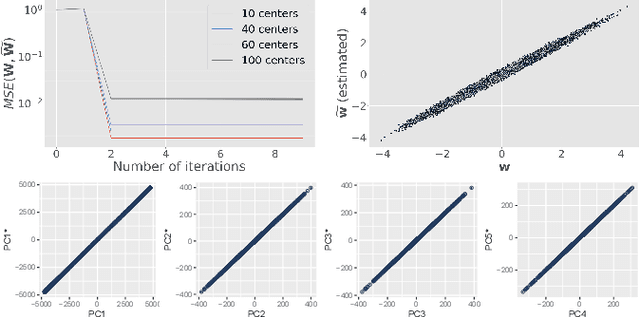
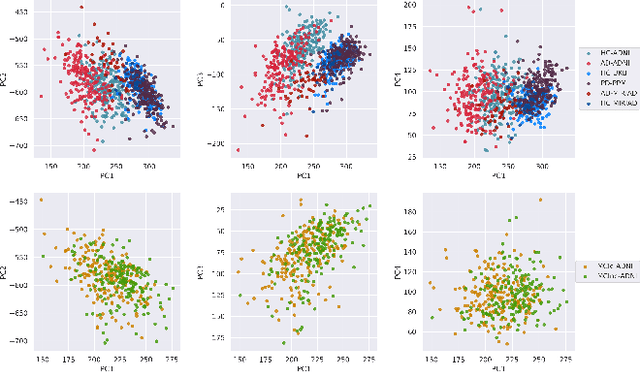
At this moment, databanks worldwide contain brain images of previously unimaginable numbers. Combined with developments in data science, these massive data provide the potential to better understand the genetic underpinnings of brain diseases. However, different datasets, which are stored at different institutions, cannot always be shared directly due to privacy and legal concerns, thus limiting the full exploitation of big data in the study of brain disorders. Here we propose a federated learning framework for securely accessing and meta-analyzing any biomedical data without sharing individual information. We illustrate our framework by investigating brain structural relationships across diseases and clinical cohorts. The framework is first tested on synthetic data and then applied to multi-centric, multi-database studies including ADNI, PPMI, MIRIAD and UK Biobank, showing the potential of the approach for further applications in distributed analysis of multi-centric cohorts