 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning to Hear by Seeing: It's Time for Vision Language Models to Understand Artistic Emotion from Sight and Sound
Nov 15, 2025Emotion understanding is critical for making Large Language Models (LLMs) more general, reliable, and aligned with humans. Art conveys emotion through the joint design of visual and auditory elements, yet most prior work is human-centered or single-modality, overlooking the emotion intentionally expressed by the artwork. Meanwhile, current Audio-Visual Language Models (AVLMs) typically require large-scale audio pretraining to endow Visual Language Models (VLMs) with hearing, which limits scalability. We present Vision Anchored Audio-Visual Emotion LLM (VAEmotionLLM), a two-stage framework that teaches a VLM to hear by seeing with limited audio pretraining and to understand emotion across modalities. In Stage 1, Vision-Guided Audio Alignment (VG-Align) distills the frozen visual pathway into a new audio pathway by aligning next-token distributions of the shared LLM on synchronized audio-video clips, enabling hearing without a large audio dataset. In Stage 2, a lightweight Cross-Modal Emotion Adapter (EmoAdapter), composed of the Emotion Enhancer and the Emotion Supervisor, injects emotion-sensitive residuals and applies emotion supervision to enhance cross-modal emotion understanding. We also construct ArtEmoBenchmark, an art-centric emotion benchmark that evaluates content and emotion understanding under audio-only, visual-only, and audio-visual inputs. VAEmotionLLM achieves state-of-the-art results on ArtEmoBenchmark, outperforming audio-only, visual-only, and audio-visual baselines. Ablations show that the proposed components are complementary.
Large-scale cervical precancerous screening via AI-assisted cytology whole slide image analysis
Jul 28, 2024



Cervical Cancer continues to be the leading gynecological malignancy, posing a persistent threat to women's health on a global scale. Early screening via cytology Whole Slide Image (WSI) diagnosis is critical to prevent this Cancer progression and improve survival rate, but pathologist's single test suffers inevitable false negative due to the immense number of cells that need to be reviewed within a WSI. Though computer-aided automated diagnostic models can serve as strong complement for pathologists, their effectiveness is hampered by the paucity of extensive and detailed annotations, coupled with the limited interpretability and robustness. These factors significantly hinder their practical applicability and reliability in clinical settings. To tackle these challenges, we develop an AI approach, which is a Scalable Technology for Robust and Interpretable Diagnosis built on Extensive data (STRIDE) of cervical cytology. STRIDE addresses the bottleneck of limited annotations by integrating patient-level labels with a small portion of cell-level labels through an end-to-end training strategy, facilitating scalable learning across extensive datasets. To further improve the robustness to real-world domain shifts of cytology slide-making and imaging, STRIDE employs color adversarial samples training that mimic staining and imaging variations. Lastly, to achieve pathologist-level interpretability for the trustworthiness in clinical settings, STRIDE can generate explanatory textual descriptions that simulates pathologists' diagnostic processes by cell image feature and textual description alignment. Conducting extensive experiments and evaluations in 183 medical centers with a dataset of 341,889 WSIs and 0.1 billion cells from cervical cytology patients, STRIDE has demonstrated a remarkable superiority over previous state-of-the-art techniques.
PathGen-1.6M: 1.6 Million Pathology Image-text Pairs Generation through Multi-agent Collaboration
Jun 28, 2024



Vision Language Models (VLMs) like CLIP have attracted substantial attention in pathology, serving as backbones for applications such as zero-shot image classification and Whole Slide Image (WSI) analysis. Additionally, they can function as vision encoders when combined with large language models (LLMs) to support broader capabilities. Current efforts to train pathology VLMs rely on pathology image-text pairs from platforms like PubMed, YouTube, and Twitter, which provide limited, unscalable data with generally suboptimal image quality. In this work, we leverage large-scale WSI datasets like TCGA to extract numerous high-quality image patches. We then train a large multimodal model to generate captions for these images, creating PathGen-1.6M, a dataset containing 1.6 million high-quality image-caption pairs. Our approach involves multiple agent models collaborating to extract representative WSI patches, generating and refining captions to obtain high-quality image-text pairs. Extensive experiments show that integrating these generated pairs with existing datasets to train a pathology-specific CLIP model, PathGen-CLIP, significantly enhances its ability to analyze pathological images, with substantial improvements across nine pathology-related zero-shot image classification tasks and three whole-slide image tasks. Furthermore, we construct 200K instruction-tuning data based on PathGen-1.6M and integrate PathGen-CLIP with the Vicuna LLM to create more powerful multimodal models through instruction tuning. Overall, we provide a scalable pathway for high-quality data generation in pathology, paving the way for next-generation general pathology models.
ADR: Attention Diversification Regularization for Mitigating Overfitting in Multiple Instance Learning based Whole Slide Image Classification
Jun 18, 2024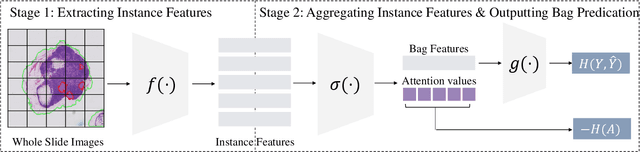

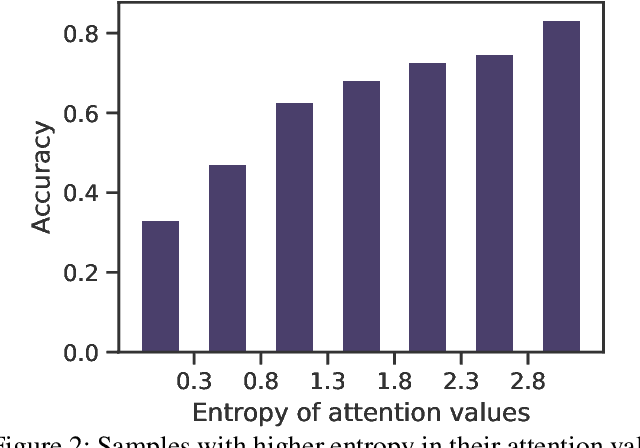
Multiple Instance Learning (MIL) has demonstrated effectiveness in analyzing whole slide images (WSIs), yet it often encounters overfitting challenges in real-world applications. This paper reveals the correlation between MIL's performance and the entropy of attention values. Based on this observation, we propose Attention Diversity Regularization (ADR), a simple but effective technique aimed at promoting high entropy in attention values. Specifically, ADR introduces a negative Shannon entropy loss for attention values into the regular MIL framework. Compared to existing methods aimed at alleviating overfitting, which often necessitate additional modules or processing steps, our ADR approach requires no such extras, demonstrating simplicity and efficiency. We evaluate our ADR on three WSI classification tasks. ADR achieves superior performance over the state-of-the-art on most of them. We also show that ADR can enhance heatmaps, aligning them better with pathologists' diagnostic criteria. The source code is available at \url{https://github.com/dazhangyu123/ADR}.
PathMMU: A Massive Multimodal Expert-Level Benchmark for Understanding and Reasoning in Pathology
Feb 05, 2024



The emergence of large multimodal models has unlocked remarkable potential in AI, particularly in pathology. However, the lack of specialized, high-quality benchmark impeded their development and precise evaluation. To address this, we introduce PathMMU, the largest and highest-quality expert-validated pathology benchmark for LMMs. It comprises 33,573 multimodal multi-choice questions and 21,599 images from various sources, and an explanation for the correct answer accompanies each question. The construction of PathMMU capitalizes on the robust capabilities of GPT-4V, utilizing approximately 30,000 gathered image-caption pairs to generate Q\&As. Significantly, to maximize PathMMU's authority, we invite six pathologists to scrutinize each question under strict standards in PathMMU's validation and test sets, while simultaneously setting an expert-level performance benchmark for PathMMU. We conduct extensive evaluations, including zero-shot assessments of 14 open-sourced and three closed-sourced LMMs and their robustness to image corruption. We also fine-tune representative LMMs to assess their adaptability to PathMMU. The empirical findings indicate that advanced LMMs struggle with the challenging PathMMU benchmark, with the top-performing LMM, GPT-4V, achieving only a 51.7\% zero-shot performance, significantly lower than the 71.4\% demonstrated by human pathologists. After fine-tuning, even open-sourced LMMs can surpass GPT-4V with a performance of over 60\%, but still fall short of the expertise shown by pathologists. We hope that the PathMMU will offer valuable insights and foster the development of more specialized, next-generation LLMs for pathology.
Benchmarking PathCLIP for Pathology Image Analysis
Jan 05, 2024
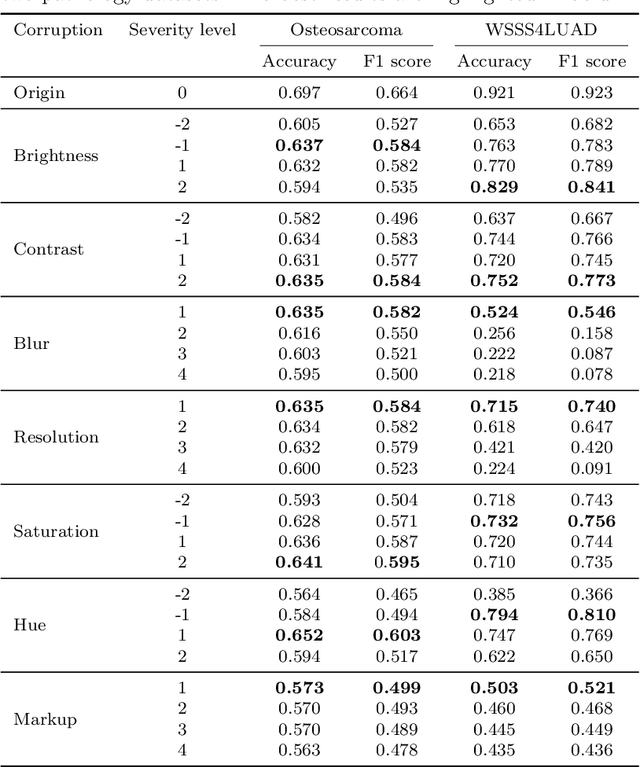

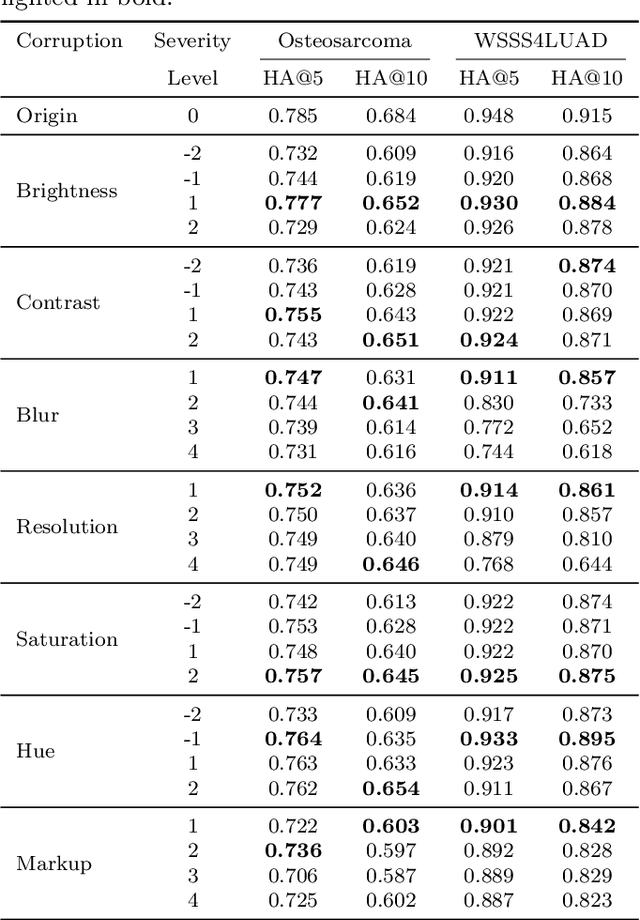
Accurate image classification and retrieval are of importance for clinical diagnosis and treatment decision-making. The recent contrastive language-image pretraining (CLIP) model has shown remarkable proficiency in understanding natural images. Drawing inspiration from CLIP, PathCLIP is specifically designed for pathology image analysis, utilizing over 200,000 image and text pairs in training. While the performance the PathCLIP is impressive, its robustness under a wide range of image corruptions remains unknown. Therefore, we conduct an extensive evaluation to analyze the performance of PathCLIP on various corrupted images from the datasets of Osteosarcoma and WSSS4LUAD. In our experiments, we introduce seven corruption types including brightness, contrast, Gaussian blur, resolution, saturation, hue, and markup at four severity levels. Through experiments, we find that PathCLIP is relatively robustness to image corruptions and surpasses OpenAI-CLIP and PLIP in zero-shot classification. Among the seven corruptions, blur and resolution can cause server performance degradation of the PathCLIP. This indicates that ensuring the quality of images is crucial before conducting a clinical test. Additionally, we assess the robustness of PathCLIP in the task of image-image retrieval, revealing that PathCLIP performs less effectively than PLIP on Osteosarcoma but performs better on WSSS4LUAD under diverse corruptions. Overall, PathCLIP presents impressive zero-shot classification and retrieval performance for pathology images, but appropriate care needs to be taken when using it. We hope this study provides a qualitative impression of PathCLIP and helps understand its differences from other CLIP models.
Masked conditional variational autoencoders for chromosome straightening
Jun 25, 2023



Karyotyping is of importance for detecting chromosomal aberrations in human disease. However, chromosomes easily appear curved in microscopic images, which prevents cytogeneticists from analyzing chromosome types. To address this issue, we propose a framework for chromosome straightening, which comprises a preliminary processing algorithm and a generative model called masked conditional variational autoencoders (MC-VAE). The processing method utilizes patch rearrangement to address the difficulty in erasing low degrees of curvature, providing reasonable preliminary results for the MC-VAE. The MC-VAE further straightens the results by leveraging chromosome patches conditioned on their curvatures to learn the mapping between banding patterns and conditions. During model training, we apply a masking strategy with a high masking ratio to train the MC-VAE with eliminated redundancy. This yields a non-trivial reconstruction task, allowing the model to effectively preserve chromosome banding patterns and structure details in the reconstructed results. Extensive experiments on three public datasets with two stain styles show that our framework surpasses the performance of state-of-the-art methods in retaining banding patterns and structure details. Compared to using real-world bent chromosomes, the use of high-quality straightened chromosomes generated by our proposed method can improve the performance of various deep learning models for chromosome classification by a large margin. Such a straightening approach has the potential to be combined with other karyotyping systems to assist cytogeneticists in chromosome analysis.
Deformable Proposal-Aware P2PNet: A Universal Network for Cell Recognition under Point Supervision
Mar 05, 2023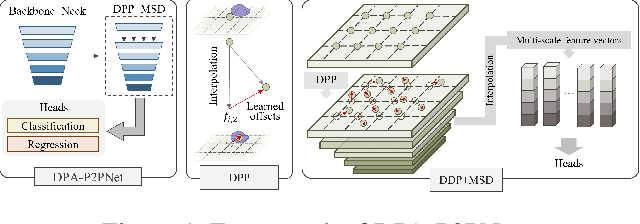

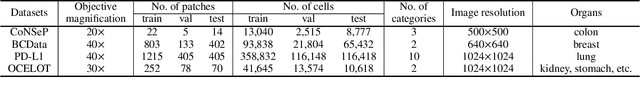
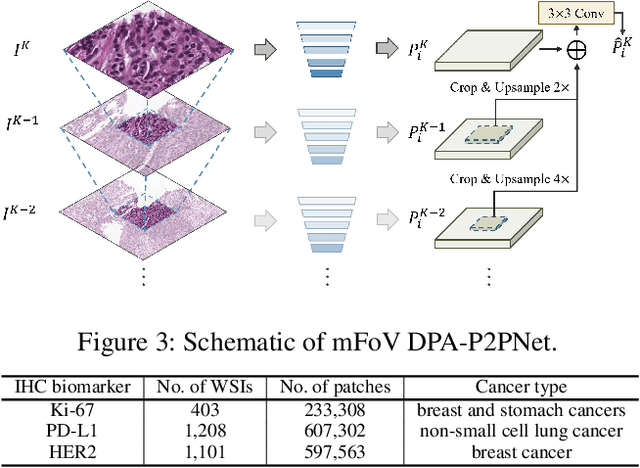
Point-based cell recognition, which aims to localize and classify cells present in a pathology image, is a fundamental task in digital pathology image analysis. The recently developed point-to-point network (P2PNet) has achieved unprecedented cell recognition accuracy and efficiency compared to methods that rely on intermediate density map representations. However, P2PNet could not leverage multi-scale information since it can only decode a single feature map. Moreover, the distribution of predefined point proposals, which is determined by data properties, restricts the resolution of the feature map to decode, i.e., the encoder design. To lift these limitations, we propose a variant of P2PNet named deformable proposal-aware P2PNet (DPA-P2PNet) in this study. The proposed method uses coordinates of point proposals to directly extract multi-scale region-of-interest (ROI) features for feature enhancement. Such a design also opens up possibilities to exploit dynamic distributions of proposals. We further devise a deformation module to improve the proposal quality. Extensive experiments on four datasets with various staining styles demonstrate that DPA-P2PNet outperforms the state-of-the-art methods on point-based cell recognition, which reveals the high potentiality in assisting pathologist assessments.
ChrSNet: Chromosome Straightening using Self-attention Guided Networks
Jul 01, 2022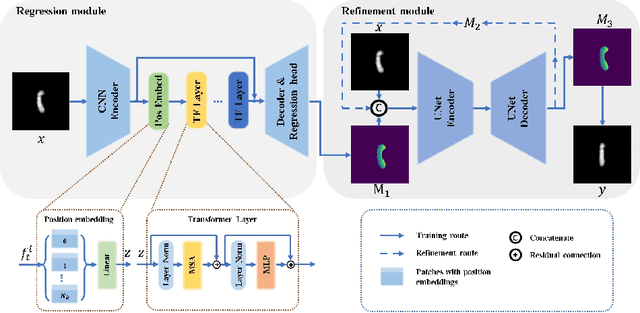
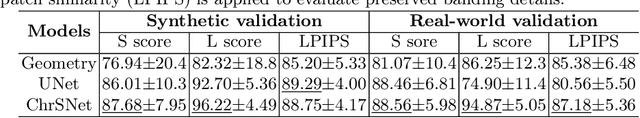
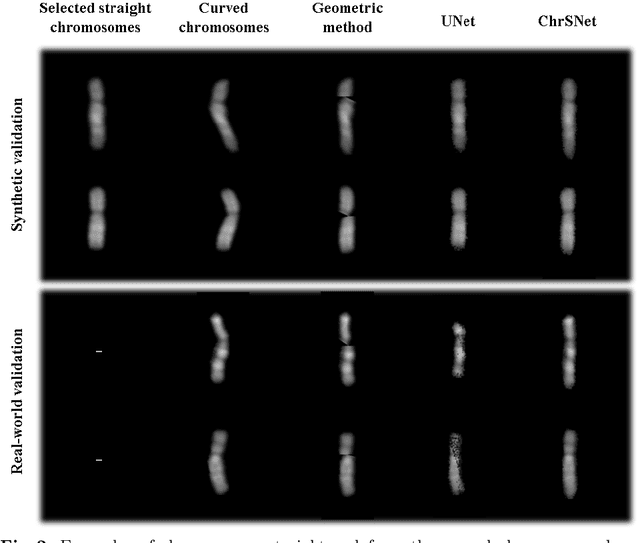
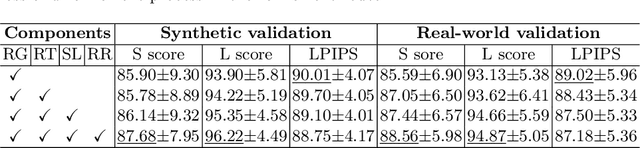
Karyotyping is an important procedure to assess the possible existence of chromosomal abnormalities. However, because of the non-rigid nature, chromosomes are usually heavily curved in microscopic images and such deformed shapes hinder the chromosome analysis for cytogeneticists. In this paper, we present a self-attention guided framework to erase the curvature of chromosomes. The proposed framework extracts spatial information and local textures to preserve banding patterns in a regression module. With complementary information from the bent chromosome, a refinement module is designed to further improve fine details. In addition, we propose two dedicated geometric constraints to maintain the length and restore the distortion of chromosomes. To train our framework, we create a synthetic dataset where curved chromosomes are generated from the real-world straight chromosomes by grid-deformation. Quantitative and qualitative experiments are conducted on synthetic and real-world data. Experimental results show that our proposed method can effectively straighten bent chromosomes while keeping banding details and length.
Dispensed Transformer Network for Unsupervised Domain Adaptation
Oct 28, 2021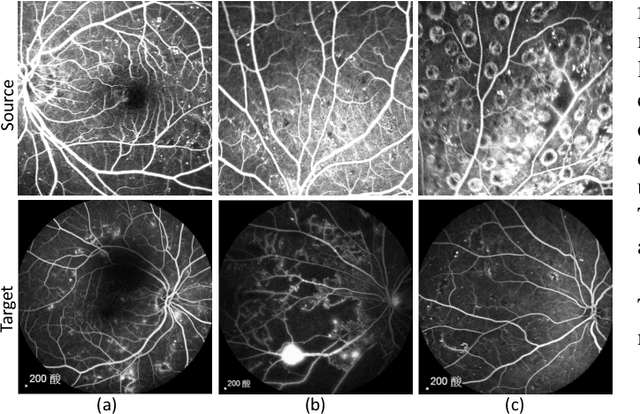
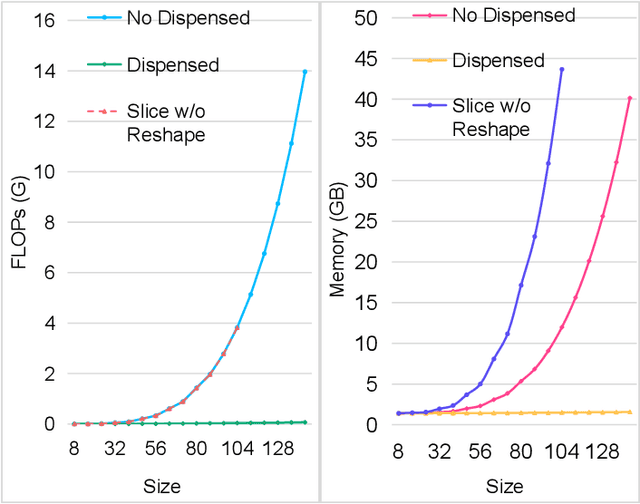
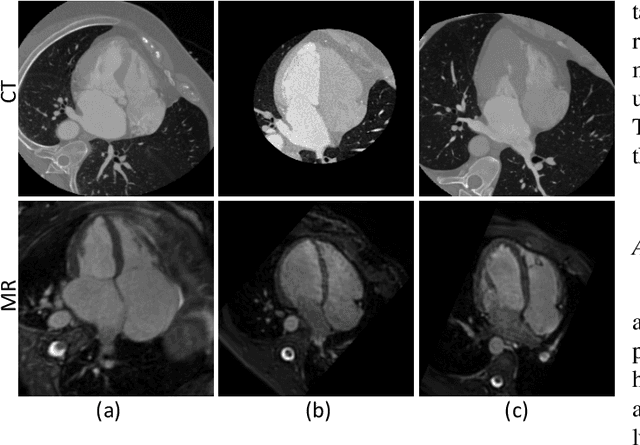
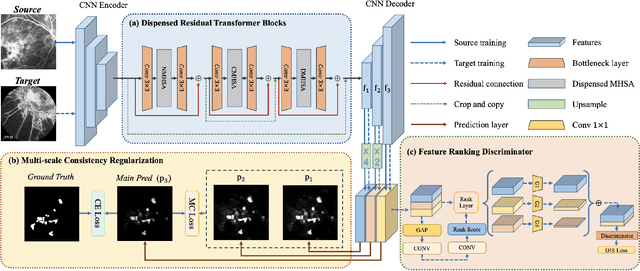
Accurate segmentation is a crucial step in medical image analysis and applying supervised machine learning to segment the organs or lesions has been substantiated effective. However, it is costly to perform data annotation that provides ground truth labels for training the supervised algorithms, and the high variance of data that comes from different domains tends to severely degrade system performance over cross-site or cross-modality datasets. To mitigate this problem, a novel unsupervised domain adaptation (UDA) method named dispensed Transformer network (DTNet) is introduced in this paper. Our novel DTNet contains three modules. First, a dispensed residual transformer block is designed, which realizes global attention by dispensed interleaving operation and deals with the excessive computational cost and GPU memory usage of the Transformer. Second, a multi-scale consistency regularization is proposed to alleviate the loss of details in the low-resolution output for better feature alignment. Finally, a feature ranking discriminator is introduced to automatically assign different weights to domain-gap features to lessen the feature distribution distance, reducing the performance shift of two domains. The proposed method is evaluated on large fluorescein angiography (FA) retinal nonperfusion (RNP) cross-site dataset with 676 images and a wide used cross-modality dataset from the MM-WHS challenge. Extensive results demonstrate that our proposed network achieves the best performance in comparison with several state-of-the-art techniques.