 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSteganographic Backdoor Attacks in NLP: Ultra-Low Poisoning and Defense Evasion
Nov 18, 2025Transformer models are foundational to natural language processing (NLP) applications, yet remain vulnerable to backdoor attacks introduced through poisoned data, which implant hidden behaviors during training. To strengthen the ability to prevent such compromises, recent research has focused on designing increasingly stealthy attacks to stress-test existing defenses, pairing backdoor behaviors with stylized artifact or token-level perturbation triggers. However, this trend diverts attention from the harder and more realistic case: making the model respond to semantic triggers such as specific names or entities, where a successful backdoor could manipulate outputs tied to real people or events in deployed systems. Motivated by this growing disconnect, we introduce SteganoBackdoor, bringing stealth techniques back into line with practical threat models. Leveraging innocuous properties from natural-language steganography, SteganoBackdoor applies a gradient-guided data optimization process to transform semantic trigger seeds into steganographic carriers that embed a high backdoor payload, remain fluent, and exhibit no representational resemblance to the trigger. Across diverse experimental settings, SteganoBackdoor achieves over 99% attack success at an order-of-magnitude lower data-poisoning rate than prior approaches while maintaining unparalleled evasion against a comprehensive suite of data-level defenses. By revealing this practical and covert attack, SteganoBackdoor highlights an urgent blind spot in current defenses and demands immediate attention to adversarial data defenses and real-world threat modeling.
Privacy-preserving Universal Adversarial Defense for Black-box Models
Aug 20, 2024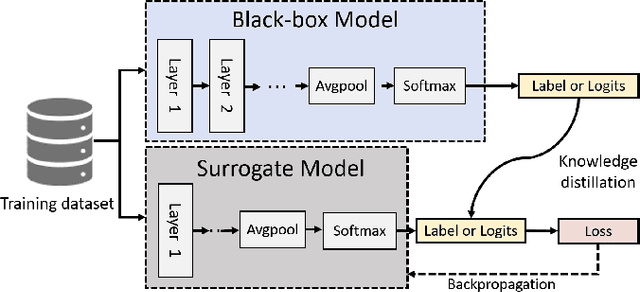
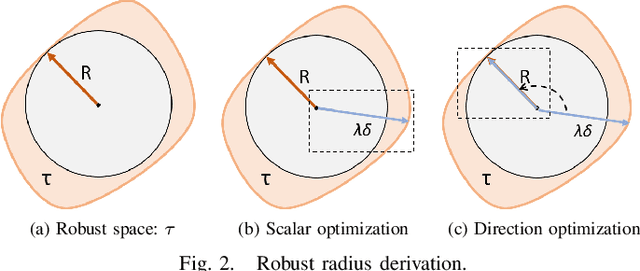
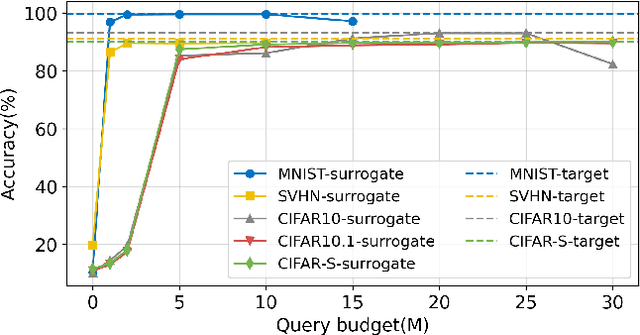
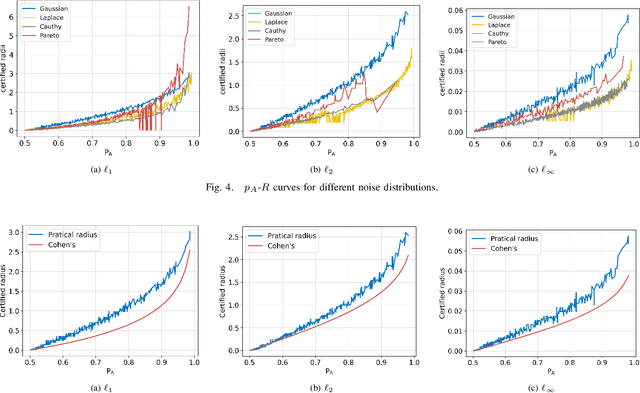
Deep neural networks (DNNs) are increasingly used in critical applications such as identity authentication and autonomous driving, where robustness against adversarial attacks is crucial. These attacks can exploit minor perturbations to cause significant prediction errors, making it essential to enhance the resilience of DNNs. Traditional defense methods often rely on access to detailed model information, which raises privacy concerns, as model owners may be reluctant to share such data. In contrast, existing black-box defense methods fail to offer a universal defense against various types of adversarial attacks. To address these challenges, we introduce DUCD, a universal black-box defense method that does not require access to the target model's parameters or architecture. Our approach involves distilling the target model by querying it with data, creating a white-box surrogate while preserving data privacy. We further enhance this surrogate model using a certified defense based on randomized smoothing and optimized noise selection, enabling robust defense against a broad range of adversarial attacks. Comparative evaluations between the certified defenses of the surrogate and target models demonstrate the effectiveness of our approach. Experiments on multiple image classification datasets show that DUCD not only outperforms existing black-box defenses but also matches the accuracy of white-box defenses, all while enhancing data privacy and reducing the success rate of membership inference attacks.
ViTime: A Visual Intelligence-Based Foundation Model for Time Series Forecasting
Jul 10, 2024



The success of large pretrained models in natural language processing (NLP) and computer vision (CV) has opened new avenues for constructing foundation models for time series forecasting (TSF). Traditional TSF foundation models rely heavily on numerical data fitting. In contrast, the human brain is inherently skilled at processing visual information, prefer predicting future trends by observing visualized sequences. From a biomimetic perspective, utilizing models to directly process numerical sequences might not be the most effective route to achieving Artificial General Intelligence (AGI). This paper proposes ViTime, a novel Visual Intelligence-based foundation model for TSF. ViTime overcomes the limitations of numerical time series data fitting by utilizing visual data processing paradigms and employs a innovative data synthesis method during training, called Real Time Series (RealTS). Experiments on a diverse set of previously unseen forecasting datasets demonstrate that ViTime achieves state-of-the-art zero-shot performance, even surpassing the best individually trained supervised models in some situations. These findings suggest that visual intelligence can significantly enhance time series analysis and forecasting, paving the way for more advanced and versatile models in the field. The code for our framework is accessible at https://github.com/IkeYang/ViTime.
DYNA: Disease-Specific Language Model for Variant Pathogenicity
May 31, 2024



Clinical variant classification of pathogenic versus benign genetic variants remains a challenge in clinical genetics. Recently, the proposition of genomic foundation models has improved the generic variant effect prediction (VEP) accuracy via weakly-supervised or unsupervised training. However, these VEPs are not disease-specific, limiting their adaptation at the point of care. To address this problem, we propose DYNA: Disease-specificity fine-tuning via a Siamese neural network broadly applicable to all genomic foundation models for more effective variant effect predictions in disease-specific contexts. We evaluate DYNA in two distinct disease-relevant tasks. For coding VEPs, we focus on various cardiovascular diseases, where gene-disease relationships of loss-of-function vs. gain-of-function dictate disease-specific VEP. For non-coding VEPs, we apply DYNA to an essential post-transcriptional regulatory axis of RNA splicing, the most common non-coding pathogenic mechanism in established clinical VEP guidelines. In both cases, DYNA fine-tunes various pre-trained genomic foundation models on small, rare variant sets. The DYNA fine-tuned models show superior performance in the held-out rare variant testing set and are further replicated in large, clinically-relevant variant annotations in ClinVAR. Thus, DYNA offers a potent disease-specific variant effect prediction method, excelling in intra-gene generalization and generalization to unseen genetic variants, making it particularly valuable for disease associations and clinical applicability.
Towards Robust Graph Incremental Learning on Evolving Graphs
Feb 20, 2024



Incremental learning is a machine learning approach that involves training a model on a sequence of tasks, rather than all tasks at once. This ability to learn incrementally from a stream of tasks is crucial for many real-world applications. However, incremental learning is a challenging problem on graph-structured data, as many graph-related problems involve prediction tasks for each individual node, known as Node-wise Graph Incremental Learning (NGIL). This introduces non-independent and non-identically distributed characteristics in the sample data generation process, making it difficult to maintain the performance of the model as new tasks are added. In this paper, we focus on the inductive NGIL problem, which accounts for the evolution of graph structure (structural shift) induced by emerging tasks. We provide a formal formulation and analysis of the problem, and propose a novel regularization-based technique called Structural-Shift-Risk-Mitigation (SSRM) to mitigate the impact of the structural shift on catastrophic forgetting of the inductive NGIL problem. We show that the structural shift can lead to a shift in the input distribution for the existing tasks, and further lead to an increased risk of catastrophic forgetting. Through comprehensive empirical studies with several benchmark datasets, we demonstrate that our proposed method, Structural-Shift-Risk-Mitigation (SSRM), is flexible and easy to adapt to improve the performance of state-of-the-art GNN incremental learning frameworks in the inductive setting.
Efficient and Scalable Fine-Tune of Language Models for Genome Understanding
Feb 12, 2024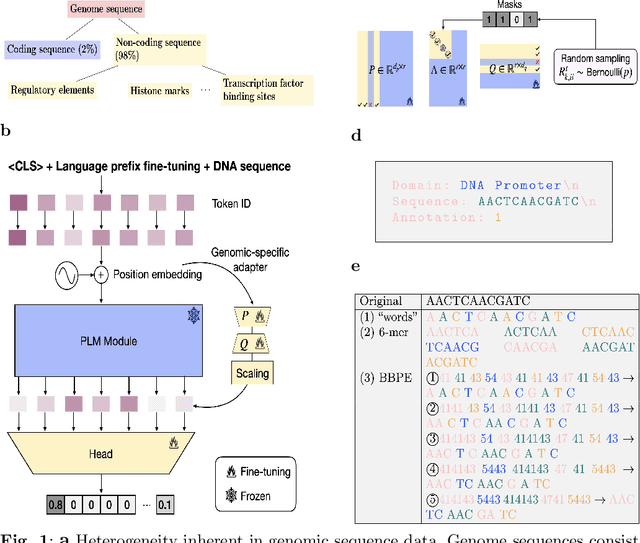
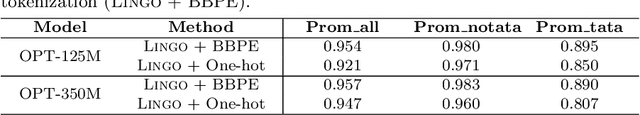
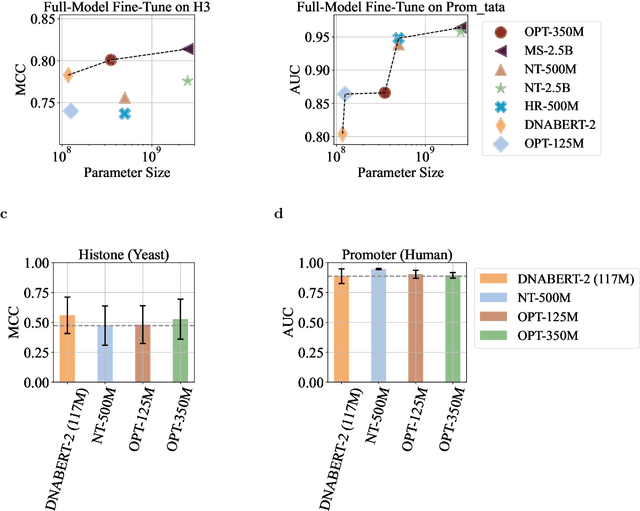
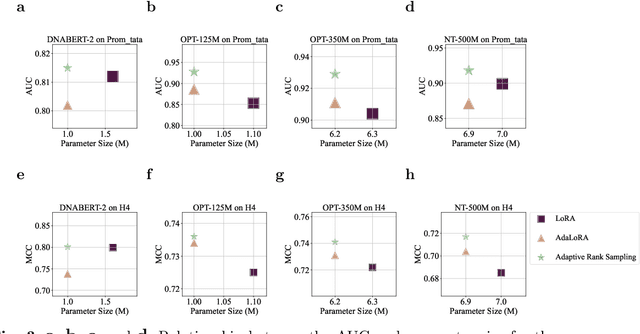
Although DNA foundation models have advanced the understanding of genomes, they still face significant challenges in the limited scale and diversity of genomic data. This limitation starkly contrasts with the success of natural language foundation models, which thrive on substantially larger scales. Furthermore, genome understanding involves numerous downstream genome annotation tasks with inherent data heterogeneity, thereby necessitating more efficient and robust fine-tuning methods tailored for genomics. Here, we present \textsc{Lingo}: \textsc{L}anguage prefix f\textsc{In}e-tuning for \textsc{G}en\textsc{O}mes. Unlike DNA foundation models, \textsc{Lingo} strategically leverages natural language foundation models' contextual cues, recalibrating their linguistic knowledge to genomic sequences. \textsc{Lingo} further accommodates numerous, heterogeneous downstream fine-tune tasks by an adaptive rank sampling method that prunes and stochastically reintroduces pruned singular vectors within small computational budgets. Adaptive rank sampling outperformed existing fine-tuning methods on all benchmarked 14 genome understanding tasks, while requiring fewer than 2\% of trainable parameters as genomic-specific adapters. Impressively, applying these adapters on natural language foundation models matched or even exceeded the performance of DNA foundation models. \textsc{Lingo} presents a new paradigm of efficient and scalable genome understanding via genomic-specific adapters on language models.
ProPath: Disease-Specific Protein Language Model for Variant Pathogenicity
Nov 08, 2023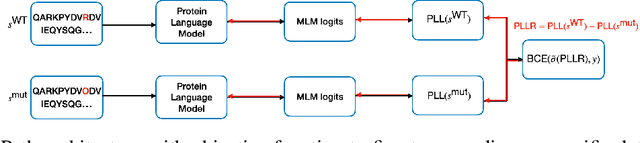
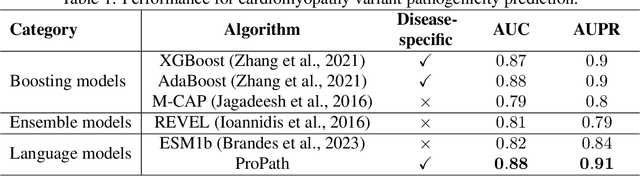
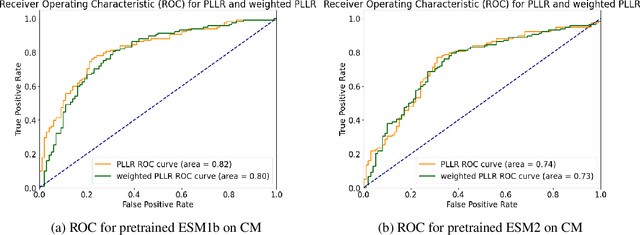
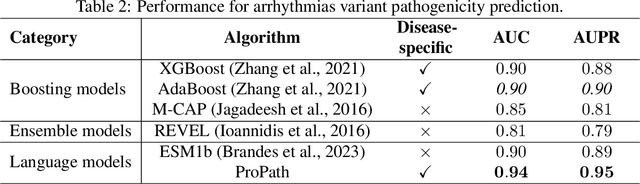
Clinical variant classification of pathogenic versus benign genetic variants remains a pivotal challenge in clinical genetics. Recently, the proposition of protein language models has improved the generic variant effect prediction (VEP) accuracy via weakly-supervised or unsupervised training. However, these VEPs are not disease-specific, limiting their adaptation at point-of-care. To address this problem, we propose a disease-specific \textsc{pro}tein language model for variant \textsc{path}ogenicity, termed ProPath, to capture the pseudo-log-likelihood ratio in rare missense variants through a siamese network. We evaluate the performance of ProPath against pre-trained language models, using clinical variant sets in inherited cardiomyopathies and arrhythmias that were not seen during training. Our results demonstrate that ProPath surpasses the pre-trained ESM1b with an over $5\%$ improvement in AUC across both datasets. Furthermore, our model achieved the highest performances across all baselines for both datasets. Thus, our ProPath offers a potent disease-specific variant effect prediction, particularly valuable for disease associations and clinical applicability.
Reusability report: Prostate cancer stratification with diverse biologically-informed neural architectures
Sep 28, 2023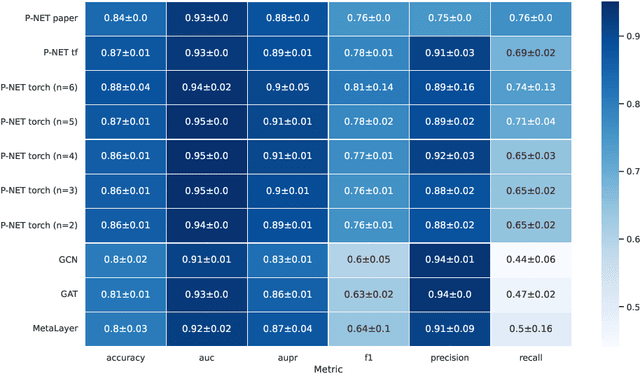
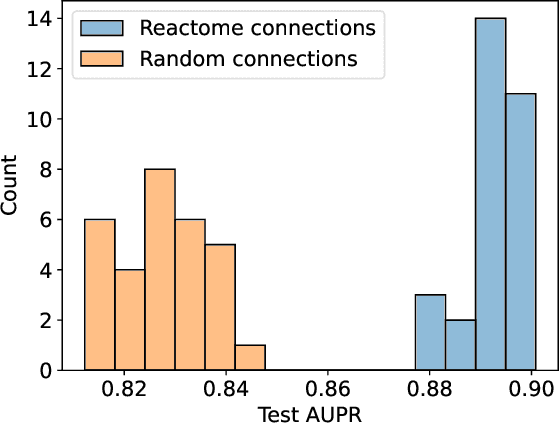
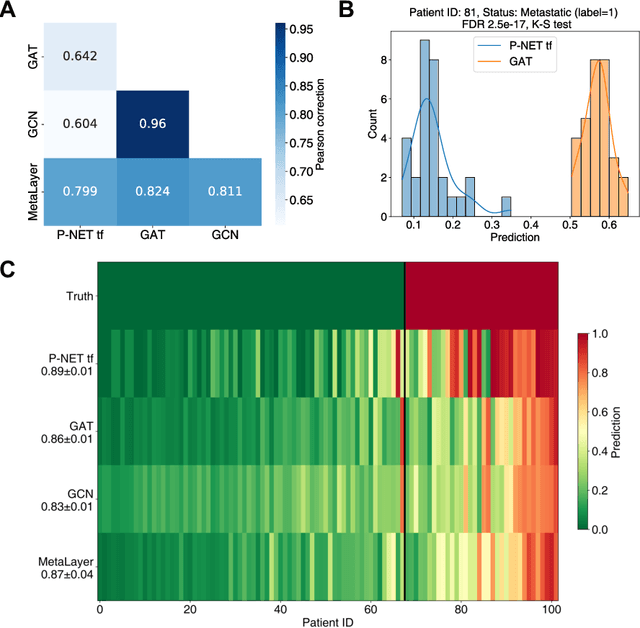
In, Elmarakeby et al., "Biologically informed deep neural network for prostate cancer discovery", a feedforward neural network with biologically informed, sparse connections (P-NET) was presented to model the state of prostate cancer. We verified the reproducibility of the study conducted by Elmarakeby et al., using both their original codebase, and our own re-implementation using more up-to-date libraries. We quantified the contribution of network sparsification by Reactome biological pathways, and confirmed its importance to P-NET's superior performance. Furthermore, we explored alternative neural architectures and approaches to incorporating biological information into the networks. We experimented with three types of graph neural networks on the same training data, and investigated the clinical prediction agreement between different models. Our analyses demonstrated that deep neural networks with distinct architectures make incorrect predictions for individual patient that are persistent across different initializations of a specific neural architecture. This suggests that different neural architectures are sensitive to different aspects of the data, an important yet under-explored challenge for clinical prediction tasks.
Interpretable neural architecture search and transfer learning for understanding sequence dependent enzymatic reactions
May 18, 2023Finely-tuned enzymatic pathways control cellular processes, and their dysregulation can lead to disease. Creating predictive and interpretable models for these pathways is challenging because of the complexity of the pathways and of the cellular and genomic contexts. Here we introduce Elektrum, a deep learning framework which addresses these challenges with data-driven and biophysically interpretable models for determining the kinetics of biochemical systems. First, it uses in vitro kinetic assays to rapidly hypothesize an ensemble of high-quality Kinetically Interpretable Neural Networks (KINNs) that predict reaction rates. It then employs a novel transfer learning step, where the KINNs are inserted as intermediary layers into deeper convolutional neural networks, fine-tuning the predictions for reaction-dependent in vivo outcomes. Elektrum makes effective use of the limited, but clean in vitro data and the noisy, yet plentiful in vivo data that captures cellular context. We apply Elektrum to predict CRISPR-Cas9 off-target editing probabilities and demonstrate that Elektrum achieves state-of-the-art performance, regularizes neural network architectures, and maintains physical interpretability.
Your time series is worth a binary image: machine vision assisted deep framework for time series forecasting
Feb 28, 2023

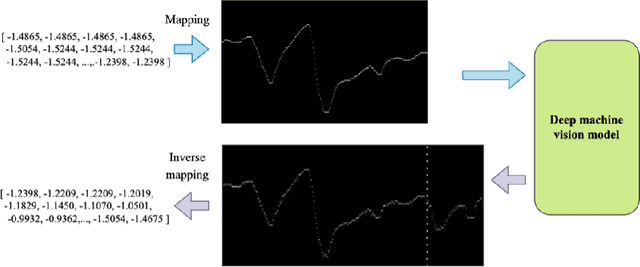

Time series forecasting (TSF) has been a challenging research area, and various models have been developed to address this task. However, almost all these models are trained with numerical time series data, which is not as effectively processed by the neural system as visual information. To address this challenge, this paper proposes a novel machine vision assisted deep time series analysis (MV-DTSA) framework. The MV-DTSA framework operates by analyzing time series data in a novel binary machine vision time series metric space, which includes a mapping and an inverse mapping function from the numerical time series space to the binary machine vision space, and a deep machine vision model designed to address the TSF task in the binary space. A comprehensive computational analysis demonstrates that the proposed MV-DTSA framework outperforms state-of-the-art deep TSF models, without requiring sophisticated data decomposition or model customization. The code for our framework is accessible at https://github.com/IkeYang/ machine-vision-assisted-deep-time-series-analysis-MV-DTSA-.