 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProgress and Opportunities of Foundation Models in Bioinformatics
Feb 06, 2024



Bioinformatics has witnessed a paradigm shift with the increasing integration of artificial intelligence (AI), particularly through the adoption of foundation models (FMs). These AI techniques have rapidly advanced, addressing historical challenges in bioinformatics such as the scarcity of annotated data and the presence of data noise. FMs are particularly adept at handling large-scale, unlabeled data, a common scenario in biological contexts due to the time-consuming and costly nature of experimentally determining labeled data. This characteristic has allowed FMs to excel and achieve notable results in various downstream validation tasks, demonstrating their ability to represent diverse biological entities effectively. Undoubtedly, FMs have ushered in a new era in computational biology, especially in the realm of deep learning. The primary goal of this survey is to conduct a systematic investigation and summary of FMs in bioinformatics, tracing their evolution, current research status, and the methodologies employed. Central to our focus is the application of FMs to specific biological problems, aiming to guide the research community in choosing appropriate FMs for their research needs. We delve into the specifics of the problem at hand including sequence analysis, structure prediction, function annotation, and multimodal integration, comparing the structures and advancements against traditional methods. Furthermore, the review analyses challenges and limitations faced by FMs in biology, such as data noise, model explainability, and potential biases. Finally, we outline potential development paths and strategies for FMs in future biological research, setting the stage for continued innovation and application in this rapidly evolving field. This comprehensive review serves not only as an academic resource but also as a roadmap for future explorations and applications of FMs in biology.
Drug Synergistic Combinations Predictions via Large-Scale Pre-Training and Graph Structure Learning
Jan 14, 2023



Drug combination therapy is a well-established strategy for disease treatment with better effectiveness and less safety degradation. However, identifying novel drug combinations through wet-lab experiments is resource intensive due to the vast combinatorial search space. Recently, computational approaches, specifically deep learning models have emerged as an efficient way to discover synergistic combinations. While previous methods reported fair performance, their models usually do not take advantage of multi-modal data and they are unable to handle new drugs or cell lines. In this study, we collected data from various datasets covering various drug-related aspects. Then, we take advantage of large-scale pre-training models to generate informative representations and features for drugs, proteins, and diseases. Based on that, a message-passing graph is built on top to propagate information together with graph structure learning flexibility. This is first introduced in the biological networks and enables us to generate pseudo-relations in the graph. Our framework achieves state-of-the-art results in comparison with other deep learning-based methods on synergistic prediction benchmark datasets. We are also capable of inferencing new drug combination data in a test on an independent set released by AstraZeneca, where 10% of improvement over previous methods is observed. In addition, we're robust against unseen drugs and surpass almost 15% AU ROC compared to the second-best model. We believe our framework contributes to both the future wet-lab discovery of novel drugs and the building of promising guidance for precise combination medicine.
A Deep Learning Approach to Generating Photospheric Vector Magnetograms of Solar Active Regions for SOHO/MDI Using SDO/HMI and BBSO Data
Nov 04, 2022Solar activity is usually caused by the evolution of solar magnetic fields. Magnetic field parameters derived from photospheric vector magnetograms of solar active regions have been used to analyze and forecast eruptive events such as solar flares and coronal mass ejections. Unfortunately, the most recent solar cycle 24 was relatively weak with few large flares, though it is the only solar cycle in which consistent time-sequence vector magnetograms have been available through the Helioseismic and Magnetic Imager (HMI) on board the Solar Dynamics Observatory (SDO) since its launch in 2010. In this paper, we look into another major instrument, namely the Michelson Doppler Imager (MDI) on board the Solar and Heliospheric Observatory (SOHO) from 1996 to 2010. The data archive of SOHO/MDI covers more active solar cycle 23 with many large flares. However, SOHO/MDI data only has line-of-sight (LOS) magnetograms. We propose a new deep learning method, named MagNet, to learn from combined LOS magnetograms, Bx and By taken by SDO/HMI along with H-alpha observations collected by the Big Bear Solar Observatory (BBSO), and to generate vector components Bx' and By', which would form vector magnetograms with observed LOS data. In this way, we can expand the availability of vector magnetograms to the period from 1996 to present. Experimental results demonstrate the good performance of the proposed method. To our knowledge, this is the first time that deep learning has been used to generate photospheric vector magnetograms of solar active regions for SOHO/MDI using SDO/HMI and H-alpha data.
E2Efold-3D: End-to-End Deep Learning Method for accurate de novo RNA 3D Structure Prediction
Jul 04, 2022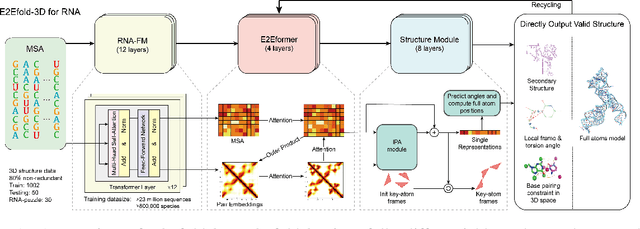
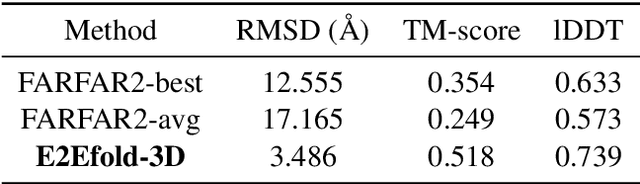
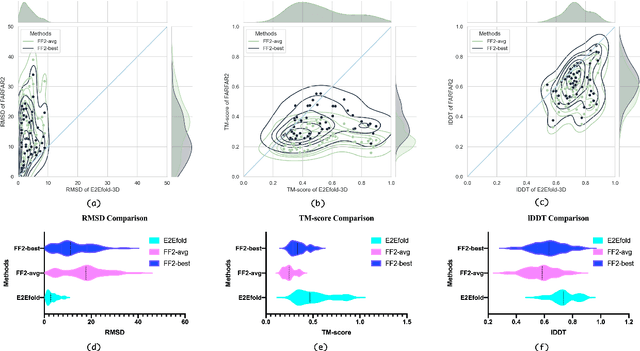
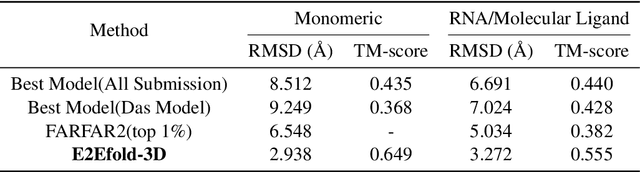
RNA structure determination and prediction can promote RNA-targeted drug development and engineerable synthetic elements design. But due to the intrinsic structural flexibility of RNAs, all the three mainstream structure determination methods (X-ray crystallography, NMR, and Cryo-EM) encounter challenges when resolving the RNA structures, which leads to the scarcity of the resolved RNA structures. Computational prediction approaches emerge as complementary to the experimental techniques. However, none of the \textit{de novo} approaches is based on deep learning since too few structures are available. Instead, most of them apply the time-consuming sampling-based strategies, and their performance seems to hit the plateau. In this work, we develop the first end-to-end deep learning approach, E2Efold-3D, to accurately perform the \textit{de novo} RNA structure prediction. Several novel components are proposed to overcome the data scarcity, such as a fully-differentiable end-to-end pipeline, secondary structure-assisted self-distillation, and parameter-efficient backbone formulation. Such designs are validated on the independent, non-overlapping RNA puzzle testing dataset and reach an average sub-4 \AA{} root-mean-square deviation, demonstrating its superior performance compared to state-of-the-art approaches. Interestingly, it also achieves promising results when predicting RNA complex structures, a feat that none of the previous systems could accomplish. When E2Efold-3D is coupled with the experimental techniques, the RNA structure prediction field can be greatly advanced.
Contrastive Cycle Adversarial Autoencoders for Single-cell Multi-omics Alignment and Integration
Dec 14, 2021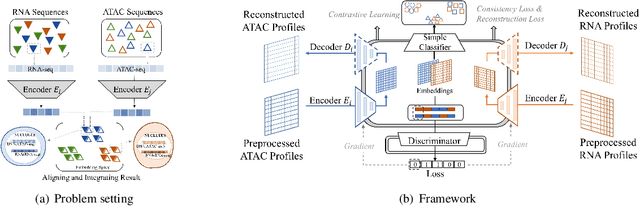
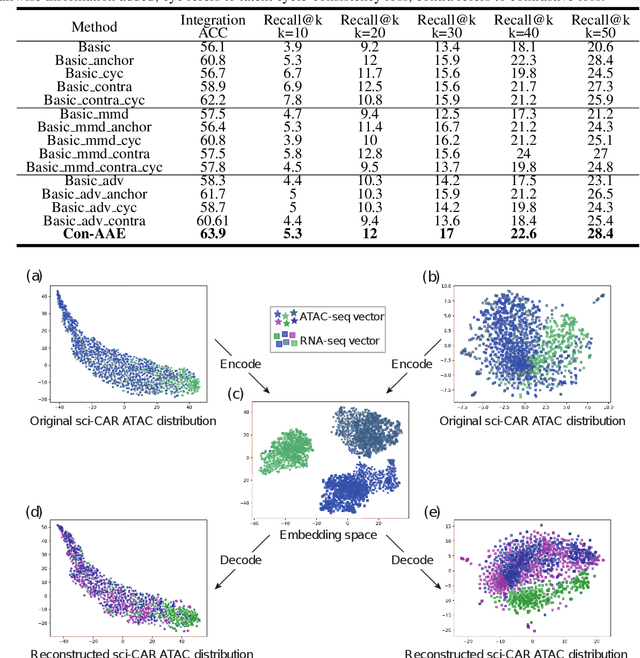
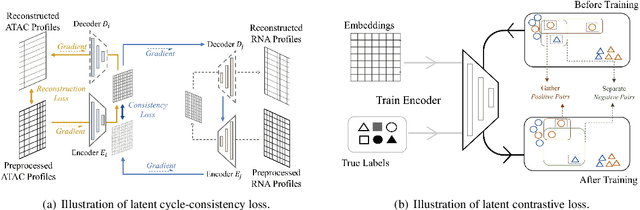
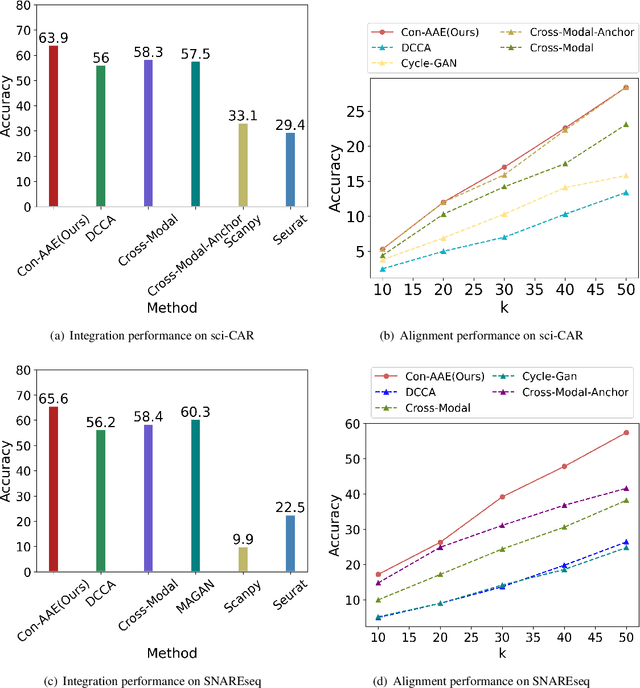
Muilti-modality data are ubiquitous in biology, especially that we have entered the multi-omics era, when we can measure the same biological object (cell) from different aspects (omics) to provide a more comprehensive insight into the cellular system. When dealing with such multi-omics data, the first step is to determine the correspondence among different modalities. In other words, we should match data from different spaces corresponding to the same object. This problem is particularly challenging in the single-cell multi-omics scenario because such data are very sparse with extremely high dimensions. Secondly, matched single-cell multi-omics data are rare and hard to collect. Furthermore, due to the limitations of the experimental environment, the data are usually highly noisy. To promote the single-cell multi-omics research, we overcome the above challenges, proposing a novel framework to align and integrate single-cell RNA-seq data and single-cell ATAC-seq data. Our approach can efficiently map the above data with high sparsity and noise from different spaces to a low-dimensional manifold in a unified space, making the downstream alignment and integration straightforward. Compared with the other state-of-the-art methods, our method performs better in both simulated and real single-cell data. The proposed method is helpful for the single-cell multi-omics research. The improvement for integration on the simulated data is significant.