 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInvestigating Data Pruning for Pretraining Biological Foundation Models at Scale
Dec 15, 2025Biological foundation models (BioFMs), pretrained on large-scale biological sequences, have recently shown strong potential in providing meaningful representations for diverse downstream bioinformatics tasks. However, such models often rely on millions to billions of training sequences and billions of parameters, resulting in prohibitive computational costs and significant barriers to reproducibility and accessibility, particularly for academic labs. To address these challenges, we investigate the feasibility of data pruning for BioFM pretraining and propose a post-hoc influence-guided data pruning framework tailored to biological domains. Our approach introduces a subset-based self-influence formulation that enables efficient estimation of sample importance at low computational cost, and builds upon it two simple yet effective selection strategies, namely Top-k Influence (Top I) and Coverage-Centric Influence (CCI). We empirically validate our method on two representative BioFMs, RNA-FM and ESM-C. For RNA, our framework consistently outperforms random selection baselines under an extreme pruning rate of over 99 percent, demonstrating its effectiveness. Furthermore, we show the generalizability of our framework on protein-related tasks using ESM-C. In particular, our coreset even outperforms random subsets that are ten times larger in both RNA and protein settings, revealing substantial redundancy in biological sequence datasets. These findings underscore the potential of influence-guided data pruning to substantially reduce the computational cost of BioFM pretraining, paving the way for more efficient, accessible, and sustainable biological AI research.
R$^2$: A LLM Based Novel-to-Screenplay Generation Framework with Causal Plot Graphs
Mar 19, 2025



Automatically adapting novels into screenplays is important for the TV, film, or opera industries to promote products with low costs. The strong performances of large language models (LLMs) in long-text generation call us to propose a LLM based framework Reader-Rewriter (R$^2$) for this task. However, there are two fundamental challenges here. First, the LLM hallucinations may cause inconsistent plot extraction and screenplay generation. Second, the causality-embedded plot lines should be effectively extracted for coherent rewriting. Therefore, two corresponding tactics are proposed: 1) A hallucination-aware refinement method (HAR) to iteratively discover and eliminate the affections of hallucinations; and 2) a causal plot-graph construction method (CPC) based on a greedy cycle-breaking algorithm to efficiently construct plot lines with event causalities. Recruiting those efficient techniques, R$^2$ utilizes two modules to mimic the human screenplay rewriting process: The Reader module adopts a sliding window and CPC to build the causal plot graphs, while the Rewriter module generates first the scene outlines based on the graphs and then the screenplays. HAR is integrated into both modules for accurate inferences of LLMs. Experimental results demonstrate the superiority of R$^2$, which substantially outperforms three existing approaches (51.3%, 22.6%, and 57.1% absolute increases) in pairwise comparison at the overall win rate for GPT-4o.
Enhancing the Protein Tertiary Structure Prediction by Multiple Sequence Alignment Generation
Jun 02, 2023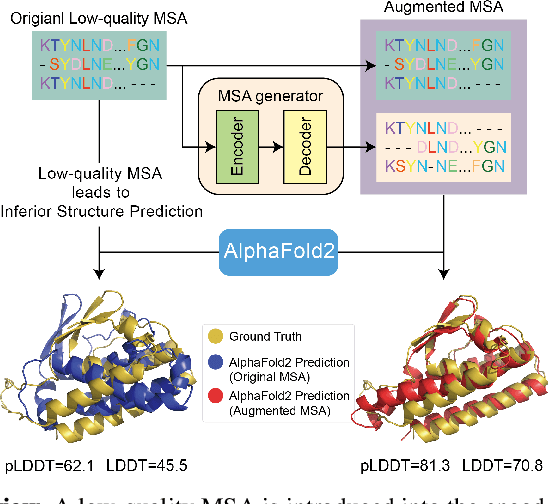



The field of protein folding research has been greatly advanced by deep learning methods, with AlphaFold2 (AF2) demonstrating exceptional performance and atomic-level precision. As co-evolution is integral to protein structure prediction, AF2's accuracy is significantly influenced by the depth of multiple sequence alignment (MSA), which requires extensive exploration of a large protein database for similar sequences. However, not all protein sequences possess abundant homologous families, and consequently, AF2's performance can degrade on such queries, at times failing to produce meaningful results. To address this, we introduce a novel generative language model, MSA-Augmenter, which leverages protein-specific attention mechanisms and large-scale MSAs to generate useful, novel protein sequences not currently found in databases. These sequences supplement shallow MSAs, enhancing the accuracy of structural property predictions. Our experiments on CASP14 demonstrate that MSA-Augmenter can generate de novo sequences that retain co-evolutionary information from inferior MSAs, thereby improving protein structure prediction quality on top of strong AF2.
E2Efold-3D: End-to-End Deep Learning Method for accurate de novo RNA 3D Structure Prediction
Jul 04, 2022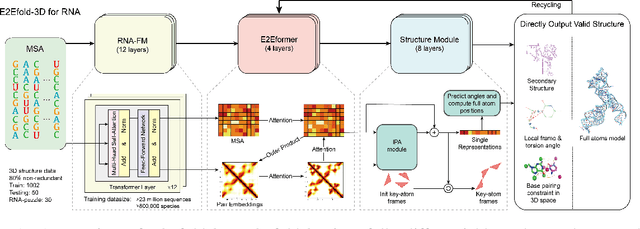
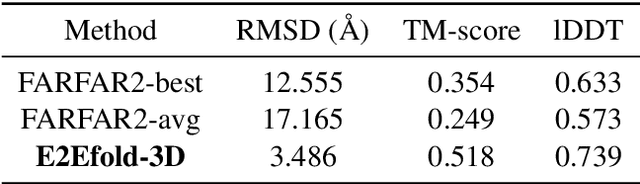
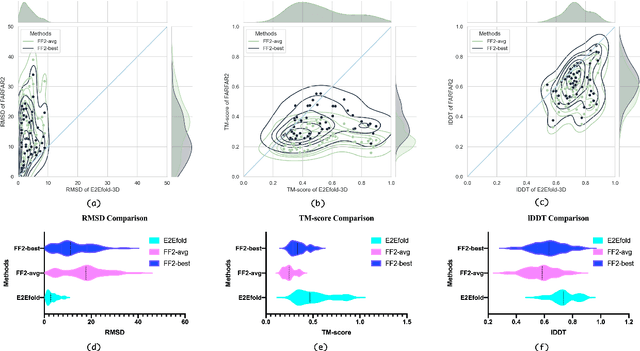
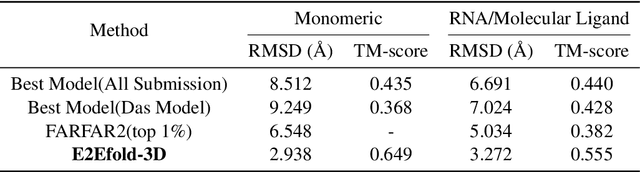
RNA structure determination and prediction can promote RNA-targeted drug development and engineerable synthetic elements design. But due to the intrinsic structural flexibility of RNAs, all the three mainstream structure determination methods (X-ray crystallography, NMR, and Cryo-EM) encounter challenges when resolving the RNA structures, which leads to the scarcity of the resolved RNA structures. Computational prediction approaches emerge as complementary to the experimental techniques. However, none of the \textit{de novo} approaches is based on deep learning since too few structures are available. Instead, most of them apply the time-consuming sampling-based strategies, and their performance seems to hit the plateau. In this work, we develop the first end-to-end deep learning approach, E2Efold-3D, to accurately perform the \textit{de novo} RNA structure prediction. Several novel components are proposed to overcome the data scarcity, such as a fully-differentiable end-to-end pipeline, secondary structure-assisted self-distillation, and parameter-efficient backbone formulation. Such designs are validated on the independent, non-overlapping RNA puzzle testing dataset and reach an average sub-4 \AA{} root-mean-square deviation, demonstrating its superior performance compared to state-of-the-art approaches. Interestingly, it also achieves promising results when predicting RNA complex structures, a feat that none of the previous systems could accomplish. When E2Efold-3D is coupled with the experimental techniques, the RNA structure prediction field can be greatly advanced.