 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnbiased organism-agnostic and highly sensitive signal peptide predictor with deep protein language model
Dec 14, 2023Signal peptide (SP) is a short peptide located in the N-terminus of proteins. It is essential to target and transfer transmembrane and secreted proteins to correct positions. Compared with traditional experimental methods to identify signal peptides, computational methods are faster and more efficient, which are more practical for analyzing thousands or even millions of protein sequences, especially for metagenomic data. Here we present Unbiased Organism-agnostic Signal Peptide Network (USPNet), a signal peptide classification and cleavage site prediction deep learning method that takes advantage of protein language models. We propose to apply label distribution-aware margin loss to handle data imbalance problems and use evolutionary information of protein to enrich representation and overcome species information dependence.
Drug Synergistic Combinations Predictions via Large-Scale Pre-Training and Graph Structure Learning
Jan 14, 2023



Drug combination therapy is a well-established strategy for disease treatment with better effectiveness and less safety degradation. However, identifying novel drug combinations through wet-lab experiments is resource intensive due to the vast combinatorial search space. Recently, computational approaches, specifically deep learning models have emerged as an efficient way to discover synergistic combinations. While previous methods reported fair performance, their models usually do not take advantage of multi-modal data and they are unable to handle new drugs or cell lines. In this study, we collected data from various datasets covering various drug-related aspects. Then, we take advantage of large-scale pre-training models to generate informative representations and features for drugs, proteins, and diseases. Based on that, a message-passing graph is built on top to propagate information together with graph structure learning flexibility. This is first introduced in the biological networks and enables us to generate pseudo-relations in the graph. Our framework achieves state-of-the-art results in comparison with other deep learning-based methods on synergistic prediction benchmark datasets. We are also capable of inferencing new drug combination data in a test on an independent set released by AstraZeneca, where 10% of improvement over previous methods is observed. In addition, we're robust against unseen drugs and surpass almost 15% AU ROC compared to the second-best model. We believe our framework contributes to both the future wet-lab discovery of novel drugs and the building of promising guidance for precise combination medicine.
HMD-AMP: Protein Language-Powered Hierarchical Multi-label Deep Forest for Annotating Antimicrobial Peptides
Nov 11, 2021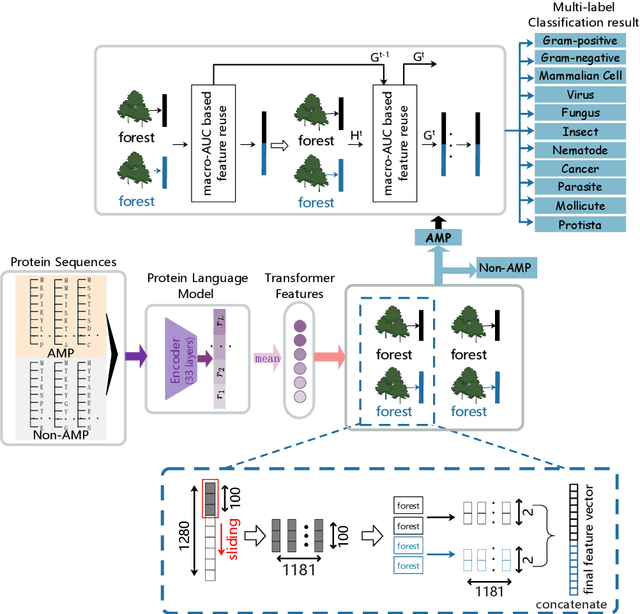

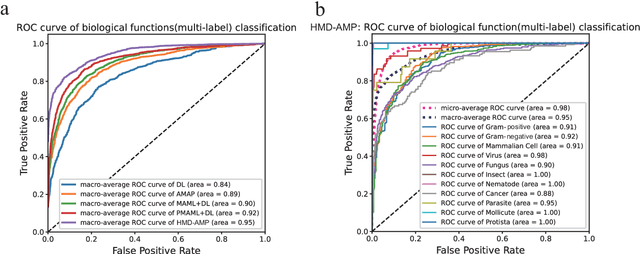
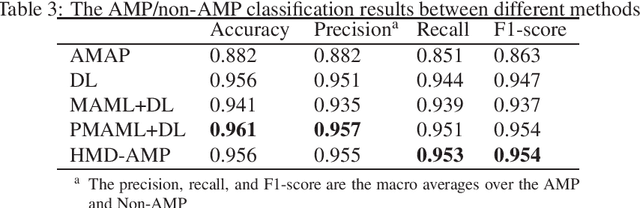
Identifying the targets of an antimicrobial peptide is a fundamental step in studying the innate immune response and combating antibiotic resistance, and more broadly, precision medicine and public health. There have been extensive studies on the statistical and computational approaches to identify (i) whether a peptide is an antimicrobial peptide (AMP) or a non-AMP and (ii) which targets are these sequences effective to (Gram-positive, Gram-negative, etc.). Despite the existing deep learning methods on this problem, most of them are unable to handle the small AMP classes (anti-insect, anti-parasite, etc.). And more importantly, some AMPs can have multiple targets, which the previous methods fail to consider. In this study, we build a diverse and comprehensive multi-label protein sequence database by collecting and cleaning amino acids from various AMP databases. To generate efficient representations and features for the small classes dataset, we take advantage of a protein language model trained on 250 million protein sequences. Based on that, we develop an end-to-end hierarchical multi-label deep forest framework, HMD-AMP, to annotate AMP comprehensively. After identifying an AMP, it further predicts what targets the AMP can effectively kill from eleven available classes. Extensive experiments suggest that our framework outperforms state-of-the-art models in both the binary classification task and the multi-label classification task, especially on the minor classes.The model is robust against reduced features and small perturbations and produces promising results. We believe HMD-AMP contributes to both the future wet-lab investigations of the innate structural properties of different antimicrobial peptides and build promising empirical underpinnings for precise medicine with antibiotics.