 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReinforcement Learning with LLM-Guided Action Spaces for Synthesizable Lead Optimization
Apr 09, 2026Lead optimization in drug discovery requires improving therapeutic properties while ensuring that proposed molecular modifications correspond to feasible synthetic routes. Existing approaches either prioritize property scores without enforcing synthesizability, or rely on expensive enumeration over large reaction networks, while direct application of Large Language Models (LLMs) frequently produces chemically invalid structures. We introduce MolReAct, a framework that formulates lead optimization as a Markov Decision Process over a synthesis-constrained action space defined by validated reaction templates. A tool-augmented LLM agent serves as a dynamic reaction environment that invokes specialized chemical analysis tools to identify reactive sites and propose chemically grounded transformations from matched templates. A policy model trained via Group Relative Policy Optimization (GRPO) selects among these constrained actions to maximize long-term oracle reward across multi-step reaction trajectories. A SMILES-based caching mechanism further reduces end-to-end optimization time by approximately 43%. Across 13 property optimization tasks from the Therapeutic Data Commons and one structure-based docking task, MolReAct achieves an average Top-10 score of 0.563, outperforming the strongest synthesizable baseline by 10.4% in relative improvement, and attains the best sample efficiency on 10 of 14 tasks. Ablations confirm that both tool-augmented reaction proposals and trajectory-level policy optimization contribute complementary gains. By grounding every step in validated reaction templates, MolReAct produces molecules that are property-improved and each accompanied by an explicit synthetic pathway.
CoSA: Compressed Sensing-Based Adaptation of Large Language Models
Feb 05, 2026Parameter-Efficient Fine-Tuning (PEFT) has emerged as a practical paradigm for adapting large language models (LLMs) without updating all parameters. Most existing approaches, such as LoRA and PiSSA, rely on low-rank decompositions of weight updates. However, the low-rank assumption may restrict expressivity, particularly in task-specific adaptation scenarios where singular values are distributed relatively uniformly. To address this limitation, we propose CoSA (Compressed Sensing-Based Adaptation), a new PEFT method extended from compressed sensing theory. Instead of constraining weight updates to a low-rank subspace, CoSA expresses them through fixed random projection matrices and a compact learnable core. We provide a formal theoretical analysis of CoSA as a synthesis process, proving that weight updates can be compactly encoded into a low-dimensional space and mapped back through random projections. Extensive experimental results show that CoSA provides a principled perspective for efficient and expressive multi-scale model adaptation. Specifically, we evaluate CoSA on 10 diverse tasks, including natural language understanding and generation, employing 5 models of different scales from RoBERTa, Llama, and Qwen families. Across these settings, CoSA consistently matches or outperforms state-of-the-art PEFT methods.
YOSO: You-Only-Sample-Once via Compressed Sensing for Graph Neural Network Training
Nov 08, 2024

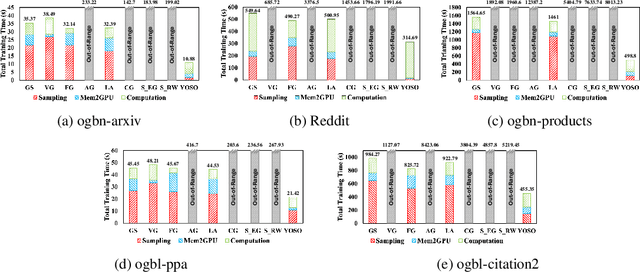

Graph neural networks (GNNs) have become essential tools for analyzing non-Euclidean data across various domains. During training stage, sampling plays an important role in reducing latency by limiting the number of nodes processed, particularly in large-scale applications. However, as the demand for better prediction performance grows, existing sampling algorithms become increasingly complex, leading to significant overhead. To mitigate this, we propose YOSO (You-Only-Sample-Once), an algorithm designed to achieve efficient training while preserving prediction accuracy. YOSO introduces a compressed sensing (CS)-based sampling and reconstruction framework, where nodes are sampled once at input layer, followed by a lossless reconstruction at the output layer per epoch. By integrating the reconstruction process with the loss function of specific learning tasks, YOSO not only avoids costly computations in traditional compressed sensing (CS) methods, such as orthonormal basis calculations, but also ensures high-probability accuracy retention which equivalent to full node participation. Experimental results on node classification and link prediction demonstrate the effectiveness and efficiency of YOSO, reducing GNN training by an average of 75\% compared to state-of-the-art methods, while maintaining accuracy on par with top-performing baselines.
MolX: Enhancing Large Language Models for Molecular Learning with A Multi-Modal Extension
Jun 10, 2024
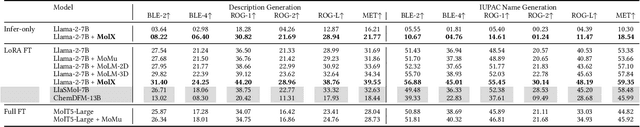
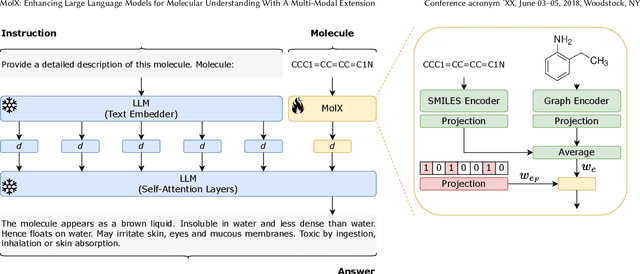
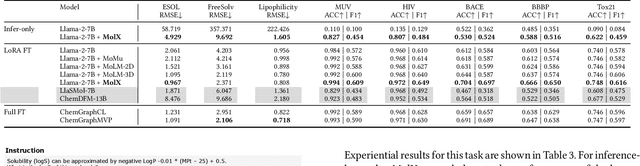
Recently, Large Language Models (LLMs) with their strong task-handling capabilities have shown remarkable advancements across a spectrum of fields, moving beyond natural language understanding. However, their proficiency within the chemistry domain remains restricted, especially in solving professional molecule-related tasks. This challenge is attributed to their inherent limitations in comprehending molecules using only common textual representations, i.e., SMILES strings. In this study, we seek to enhance the ability of LLMs to comprehend molecules by designing and equipping them with a multi-modal external module, namely MolX. In particular, instead of directly using a SMILES string to represent a molecule, we utilize specific encoders to extract fine-grained features from both SMILES string and 2D molecular graph representations for feeding into an LLM. Moreover, a human-defined molecular fingerprint is incorporated to leverage its embedded domain knowledge. Then, to establish an alignment between MolX and the LLM's textual input space, the whole model in which the LLM is frozen, is pre-trained with a versatile strategy including a diverse set of tasks. Extensive experimental evaluations demonstrate that our proposed method only introduces a small number of trainable parameters while outperforming baselines on various downstream molecule-related tasks ranging from molecule-to-text translation to retrosynthesis, with and without fine-tuning the LLM.
You do not have to train Graph Neural Networks at all on text-attributed graphs
Apr 17, 2024
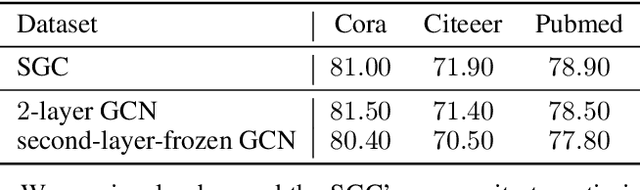
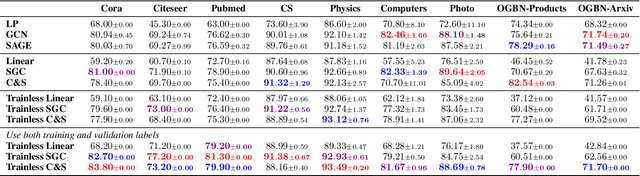
Graph structured data, specifically text-attributed graphs (TAG), effectively represent relationships among varied entities. Such graphs are essential for semi-supervised node classification tasks. Graph Neural Networks (GNNs) have emerged as a powerful tool for handling this graph-structured data. Although gradient descent is commonly utilized for training GNNs for node classification, this study ventures into alternative methods, eliminating the iterative optimization processes. We introduce TrainlessGNN, a linear GNN model capitalizing on the observation that text encodings from the same class often cluster together in a linear subspace. This model constructs a weight matrix to represent each class's node attribute subspace, offering an efficient approach to semi-supervised node classification on TAG. Extensive experiments reveal that our trainless models can either match or even surpass their conventionally trained counterparts, demonstrating the possibility of refraining from gradient descent in certain configurations.
CORE: Data Augmentation for Link Prediction via Information Bottleneck
Apr 17, 2024
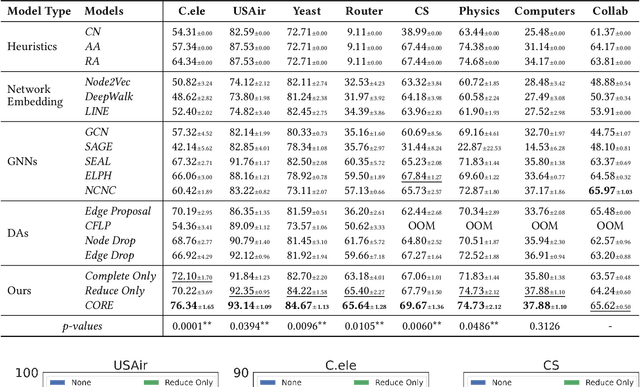

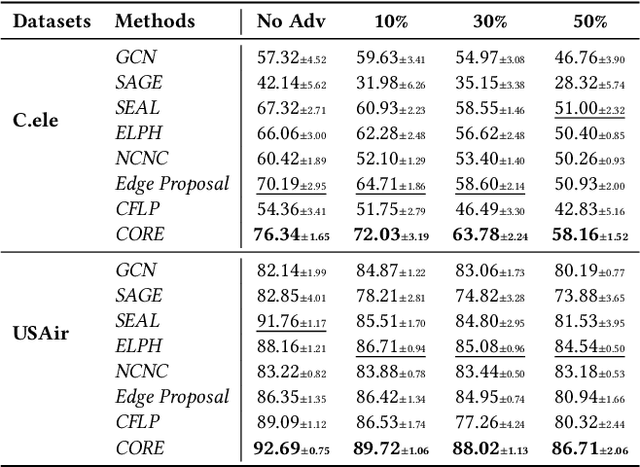
Link prediction (LP) is a fundamental task in graph representation learning, with numerous applications in diverse domains. However, the generalizability of LP models is often compromised due to the presence of noisy or spurious information in graphs and the inherent incompleteness of graph data. To address these challenges, we draw inspiration from the Information Bottleneck principle and propose a novel data augmentation method, COmplete and REduce (CORE) to learn compact and predictive augmentations for LP models. In particular, CORE aims to recover missing edges in graphs while simultaneously removing noise from the graph structures, thereby enhancing the model's robustness and performance. Extensive experiments on multiple benchmark datasets demonstrate the applicability and superiority of CORE over state-of-the-art methods, showcasing its potential as a leading approach for robust LP in graph representation learning.
Improving Out-of-Vocabulary Handling in Recommendation Systems
Mar 27, 2024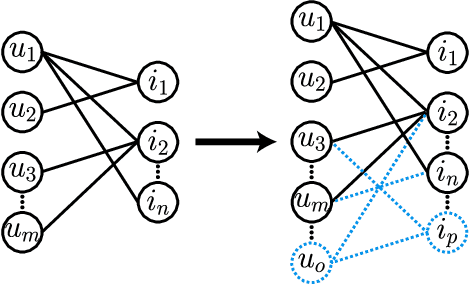


Recommendation systems (RS) are an increasingly relevant area for both academic and industry researchers, given their widespread impact on the daily online experiences of billions of users. One common issue in real RS is the cold-start problem, where users and items may not contain enough information to produce high-quality recommendations. This work focuses on a complementary problem: recommending new users and items unseen (out-of-vocabulary, or OOV) at training time. This setting is known as the inductive setting and is especially problematic for factorization-based models, which rely on encoding only those users/items seen at training time with fixed parameter vectors. Many existing solutions applied in practice are often naive, such as assigning OOV users/items to random buckets. In this work, we tackle this problem and propose approaches that better leverage available user/item features to improve OOV handling at the embedding table level. We discuss general-purpose plug-and-play approaches that are easily applicable to most RS models and improve inductive performance without negatively impacting transductive model performance. We extensively evaluate 9 OOV embedding methods on 5 models across 4 datasets (spanning different domains). One of these datasets is a proprietary production dataset from a prominent RS employed by a large social platform serving hundreds of millions of daily active users. In our experiments, we find that several proposed methods that exploit feature similarity using LSH consistently outperform alternatives on most model-dataset combinations, with the best method showing a mean improvement of 3.74% over the industry standard baseline in inductive performance. We release our code and hope our work helps practitioners make more informed decisions when handling OOV for their RS and further inspires academic research into improving OOV support in RS.
Universal Link Predictor By In-Context Learning on Graphs
Feb 15, 2024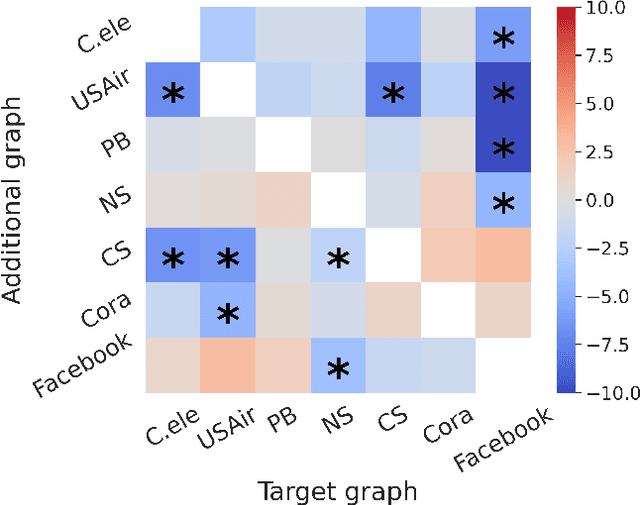
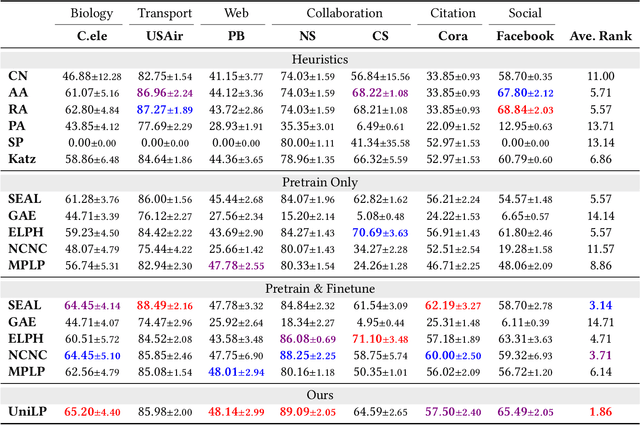
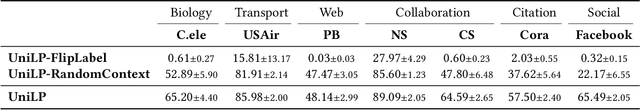
Link prediction is a crucial task in graph machine learning, where the goal is to infer missing or future links within a graph. Traditional approaches leverage heuristic methods based on widely observed connectivity patterns, offering broad applicability and generalizability without the need for model training. Despite their utility, these methods are limited by their reliance on human-derived heuristics and lack the adaptability of data-driven approaches. Conversely, parametric link predictors excel in automatically learning the connectivity patterns from data and achieving state-of-the-art but fail short to directly transfer across different graphs. Instead, it requires the cost of extensive training and hyperparameter optimization to adapt to the target graph. In this work, we introduce the Universal Link Predictor (UniLP), a novel model that combines the generalizability of heuristic approaches with the pattern learning capabilities of parametric models. UniLP is designed to autonomously identify connectivity patterns across diverse graphs, ready for immediate application to any unseen graph dataset without targeted training. We address the challenge of conflicting connectivity patterns-arising from the unique distributions of different graphs-through the implementation of In-context Learning (ICL). This approach allows UniLP to dynamically adjust to various target graphs based on contextual demonstrations, thereby avoiding negative transfer. Through rigorous experimentation, we demonstrate UniLP's effectiveness in adapting to new, unseen graphs at test time, showcasing its ability to perform comparably or even outperform parametric models that have been finetuned for specific datasets. Our findings highlight UniLP's potential to set a new standard in link prediction, combining the strengths of heuristic and parametric methods in a single, versatile framework.
Node Duplication Improves Cold-start Link Prediction
Feb 15, 2024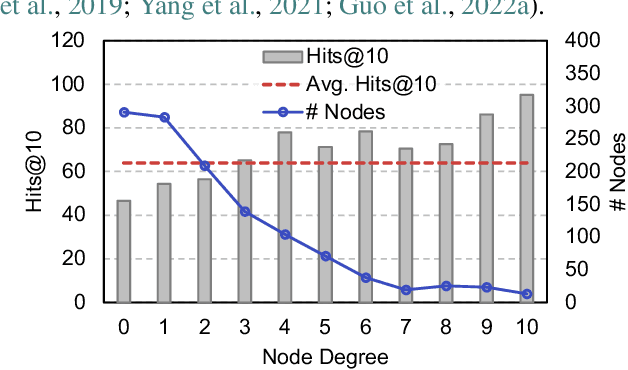
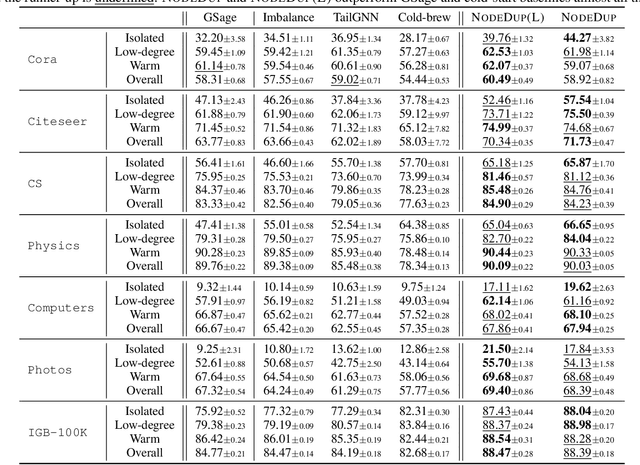
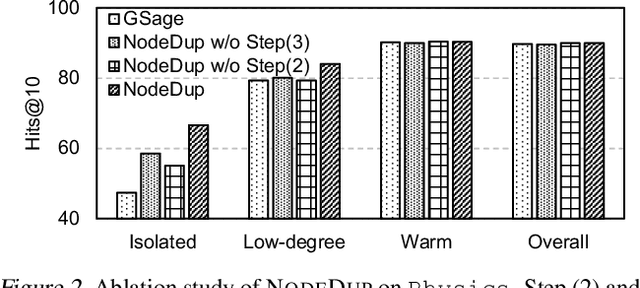
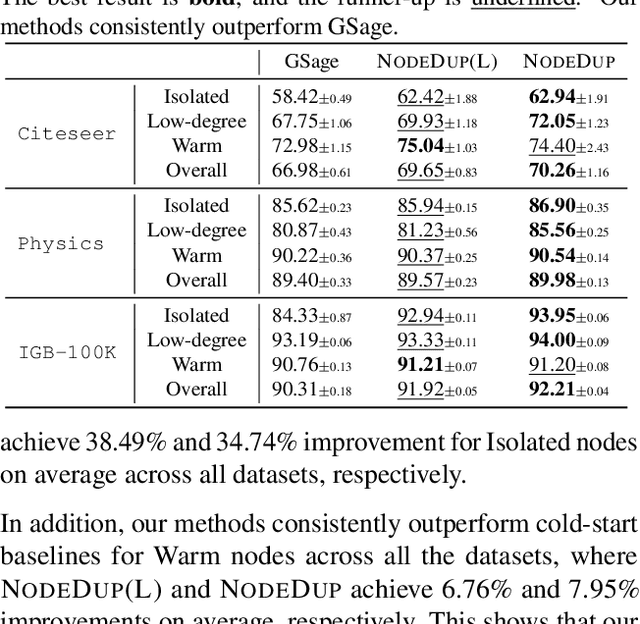
Graph Neural Networks (GNNs) are prominent in graph machine learning and have shown state-of-the-art performance in Link Prediction (LP) tasks. Nonetheless, recent studies show that GNNs struggle to produce good results on low-degree nodes despite their overall strong performance. In practical applications of LP, like recommendation systems, improving performance on low-degree nodes is critical, as it amounts to tackling the cold-start problem of improving the experiences of users with few observed interactions. In this paper, we investigate improving GNNs' LP performance on low-degree nodes while preserving their performance on high-degree nodes and propose a simple yet surprisingly effective augmentation technique called NodeDup. Specifically, NodeDup duplicates low-degree nodes and creates links between nodes and their own duplicates before following the standard supervised LP training scheme. By leveraging a ''multi-view'' perspective for low-degree nodes, NodeDup shows significant LP performance improvements on low-degree nodes without compromising any performance on high-degree nodes. Additionally, as a plug-and-play augmentation module, NodeDup can be easily applied to existing GNNs with very light computational cost. Extensive experiments show that NodeDup achieves 38.49%, 13.34%, and 6.76% improvements on isolated, low-degree, and warm nodes, respectively, on average across all datasets compared to GNNs and state-of-the-art cold-start methods.
Pure Message Passing Can Estimate Common Neighbor for Link Prediction
Sep 02, 2023



Message Passing Neural Networks (MPNNs) have emerged as the {\em de facto} standard in graph representation learning. However, when it comes to link prediction, they often struggle, surpassed by simple heuristics such as Common Neighbor (CN). This discrepancy stems from a fundamental limitation: while MPNNs excel in node-level representation, they stumble with encoding the joint structural features essential to link prediction, like CN. To bridge this gap, we posit that, by harnessing the orthogonality of input vectors, pure message-passing can indeed capture joint structural features. Specifically, we study the proficiency of MPNNs in approximating CN heuristics. Based on our findings, we introduce the Message Passing Link Predictor (MPLP), a novel link prediction model. MPLP taps into quasi-orthogonal vectors to estimate link-level structural features, all while preserving the node-level complexities. Moreover, our approach demonstrates that leveraging message-passing to capture structural features could offset MPNNs' expressiveness limitations at the expense of estimation variance. We conduct experiments on benchmark datasets from various domains, where our method consistently outperforms the baseline methods.