 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHow Close Are We? Limitations and Progress of AI Models in Banff Lesion Scoring
Oct 31, 2025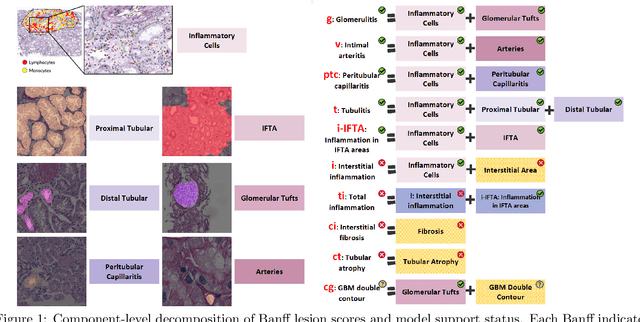
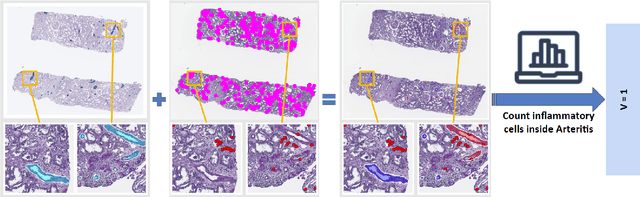
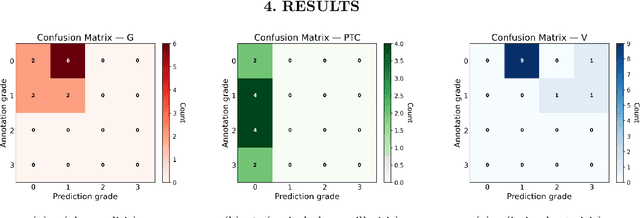
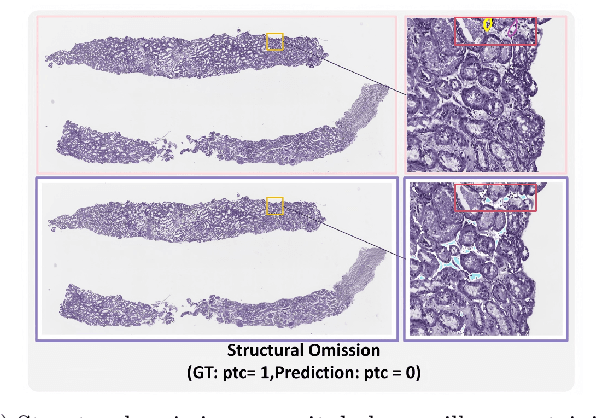
The Banff Classification provides the global standard for evaluating renal transplant biopsies, yet its semi-quantitative nature, complex criteria, and inter-observer variability present significant challenges for computational replication. In this study, we explore the feasibility of approximating Banff lesion scores using existing deep learning models through a modular, rule-based framework. We decompose each Banff indicator - such as glomerulitis (g), peritubular capillaritis (ptc), and intimal arteritis (v) - into its constituent structural and inflammatory components, and assess whether current segmentation and detection tools can support their computation. Model outputs are mapped to Banff scores using heuristic rules aligned with expert guidelines, and evaluated against expert-annotated ground truths. Our findings highlight both partial successes and critical failure modes, including structural omission, hallucination, and detection ambiguity. Even when final scores match expert annotations, inconsistencies in intermediate representations often undermine interpretability. These results reveal the limitations of current AI pipelines in replicating computational expert-level grading, and emphasize the importance of modular evaluation and computational Banff grading standard in guiding future model development for transplant pathology.
Quantitative Benchmarking of Anomaly Detection Methods in Digital Pathology
Jun 24, 2025Anomaly detection has been widely studied in the context of industrial defect inspection, with numerous methods developed to tackle a range of challenges. In digital pathology, anomaly detection holds significant potential for applications such as rare disease identification, artifact detection, and biomarker discovery. However, the unique characteristics of pathology images, such as their large size, multi-scale structures, stain variability, and repetitive patterns, introduce new challenges that current anomaly detection algorithms struggle to address. In this quantitative study, we benchmark over 20 classical and prevalent anomaly detection methods through extensive experiments. We curated five digital pathology datasets, both real and synthetic, to systematically evaluate these approaches. Our experiments investigate the influence of image scale, anomaly pattern types, and training epoch selection strategies on detection performance. The results provide a detailed comparison of each method's strengths and limitations, establishing a comprehensive benchmark to guide future research in anomaly detection for digital pathology images.
MagNet: Multi-Level Attention Graph Network for Predicting High-Resolution Spatial Transcriptomics
Feb 28, 2025



The rapid development of spatial transcriptomics (ST) offers new opportunities to explore the gene expression patterns within the spatial microenvironment. Current research integrates pathological images to infer gene expression, addressing the high costs and time-consuming processes to generate spatial transcriptomics data. However, as spatial transcriptomics resolution continues to improve, existing methods remain primarily focused on gene expression prediction at low-resolution spot levels. These methods face significant challenges, especially the information bottleneck, when they are applied to high-resolution HD data. To bridge this gap, this paper introduces MagNet, a multi-level attention graph network designed for accurate prediction of high-resolution HD data. MagNet employs cross-attention layers to integrate features from multi-resolution image patches hierarchically and utilizes a GAT-Transformer module to aggregate neighborhood information. By integrating multilevel features, MagNet overcomes the limitations posed by low-resolution inputs in predicting high-resolution gene expression. We systematically evaluated MagNet and existing ST prediction models on both a private spatial transcriptomics dataset and a public dataset at three different resolution levels. The results demonstrate that MagNet achieves state-of-the-art performance at both spot level and high-resolution bin levels, providing a novel methodology and benchmark for future research and applications in high-resolution HD-level spatial transcriptomics. Code is available at https://github.com/Junchao-Zhu/MagNet.
Glo-In-One-v2: Holistic Identification of Glomerular Cells, Tissues, and Lesions in Human and Mouse Histopathology
Nov 25, 2024Segmenting glomerular intraglomerular tissue and lesions traditionally depends on detailed morphological evaluations by expert nephropathologists, a labor-intensive process susceptible to interobserver variability. Our group previously developed the Glo-In-One toolkit for integrated detection and segmentation of glomeruli. In this study, we leverage the Glo-In-One toolkit to version 2 with fine-grained segmentation capabilities, curating 14 distinct labels for tissue regions, cells, and lesions across a dataset of 23,529 annotated glomeruli across human and mouse histopathology data. To our knowledge, this dataset is among the largest of its kind to date.In this study, we present a single dynamic head deep learning architecture designed to segment 14 classes within partially labeled images of human and mouse pathology data. Our model was trained using a training set derived from 368 annotated kidney whole-slide images (WSIs) to identify 5 key intraglomerular tissues covering Bowman's capsule, glomerular tuft, mesangium, mesangial cells, and podocytes. Additionally, the network segments 9 glomerular lesion classes including adhesion, capsular drop, global sclerosis, hyalinosis, mesangial lysis, microaneurysm, nodular sclerosis, mesangial expansion, and segmental sclerosis. The glomerulus segmentation model achieved a decent performance compared with baselines, and achieved a 76.5 % average Dice Similarity Coefficient (DSC). Additional, transfer learning from rodent to human for glomerular lesion segmentation model has enhanced the average segmentation accuracy across different types of lesions by more than 3 %, as measured by Dice scores. The Glo-In-One-v2 model and trained weight have been made publicly available at https: //github.com/hrlblab/Glo-In-One_v2.
Cross-Species Data Integration for Enhanced Layer Segmentation in Kidney Pathology
Aug 17, 2024Accurate delineation of the boundaries between the renal cortex and medulla is crucial for subsequent functional structural analysis and disease diagnosis. Training high-quality deep-learning models for layer segmentation relies on the availability of large amounts of annotated data. However, due to the patient's privacy of medical data and scarce clinical cases, constructing pathological datasets from clinical sources is relatively difficult and expensive. Moreover, using external natural image datasets introduces noise during the domain generalization process. Cross-species homologous data, such as mouse kidney data, which exhibits high structural and feature similarity to human kidneys, has the potential to enhance model performance on human datasets. In this study, we incorporated the collected private Periodic Acid-Schiff (PAS) stained mouse kidney dataset into the human kidney dataset for joint training. The results showed that after introducing cross-species homologous data, the semantic segmentation models based on CNN and Transformer architectures achieved an average increase of 1.77% and 1.24% in mIoU, and 1.76% and 0.89% in Dice score for the human renal cortex and medulla datasets, respectively. This approach is also capable of enhancing the model's generalization ability. This indicates that cross-species homologous data, as a low-noise trainable data source, can help improve model performance under conditions of limited clinical samples. Code is available at https://github.com/hrlblab/layer_segmentation.
Adapting Mouse Pathological Model to Human Glomerular Lesion Segmentation
Jul 25, 2024Moving from animal models to human applications in preclinical research encompasses a broad spectrum of disciplines in medical science. A fundamental element in the development of new drugs, treatments, diagnostic methods, and in deepening our understanding of disease processes is the accurate measurement of kidney tissues. Past studies have demonstrated the viability of translating glomeruli segmentation techniques from mouse models to human applications. Yet, these investigations tend to neglect the complexities involved in segmenting pathological glomeruli affected by different lesions. Such lesions present a wider range of morphological variations compared to healthy glomerular tissue, which are arguably more valuable than normal glomeruli in clinical practice. Furthermore, data on lesions from animal models can be more readily scaled up from disease models and whole kidney biopsies. This brings up a question: ``\textit{Can a pathological segmentation model trained on mouse models be effectively applied to human patients?}" To answer this question, we introduced GLAM, a deep learning study for fine-grained segmentation of human kidney lesions using a mouse model, addressing mouse-to-human transfer learning, by evaluating different learning strategies for segmenting human pathological lesions using zero-shot transfer learning and hybrid learning by leveraging mouse samples. From the results, the hybrid learning model achieved superior performance.
Circle Representation for Medical Instance Object Segmentation
Mar 18, 2024



Recently, circle representation has been introduced for medical imaging, designed specifically to enhance the detection of instance objects that are spherically shaped (e.g., cells, glomeruli, and nuclei). Given its outstanding effectiveness in instance detection, it is compelling to consider the application of circle representation for segmenting instance medical objects. In this study, we introduce CircleSnake, a simple end-to-end segmentation approach that utilizes circle contour deformation for segmenting ball-shaped medical objects at the instance level. The innovation of CircleSnake lies in these three areas: (1) It substitutes the complex bounding box-to-octagon contour transformation with a more consistent and rotation-invariant bounding circle-to-circle contour adaptation. This adaptation specifically targets ball-shaped medical objects. (2) The circle representation employed in CircleSnake significantly reduces the degrees of freedom to two, compared to eight in the octagon representation. This reduction enhances both the robustness of the segmentation performance and the rotational consistency of the method. (3) CircleSnake is the first end-to-end deep instance segmentation pipeline to incorporate circle representation, encompassing consistent circle detection, circle contour proposal, and circular convolution in a unified framework. This integration is achieved through the novel application of circular graph convolution within the context of circle detection and instance segmentation. In practical applications, such as the detection of glomeruli, nuclei, and eosinophils in pathological images, CircleSnake has demonstrated superior performance and greater rotation invariance when compared to benchmarks. The code has been made publicly available: https://github.com/hrlblab/CircleSnake.
Eosinophils Instance Object Segmentation on Whole Slide Imaging Using Multi-label Circle Representation
Aug 17, 2023Eosinophilic esophagitis (EoE) is a chronic and relapsing disease characterized by esophageal inflammation. Symptoms of EoE include difficulty swallowing, food impaction, and chest pain which significantly impact the quality of life, resulting in nutritional impairments, social limitations, and psychological distress. The diagnosis of EoE is typically performed with a threshold (15 to 20) of eosinophils (Eos) per high-power field (HPF). Since the current counting process of Eos is a resource-intensive process for human pathologists, automatic methods are desired. Circle representation has been shown as a more precise, yet less complicated, representation for automatic instance cell segmentation such as CircleSnake approach. However, the CircleSnake was designed as a single-label model, which is not able to deal with multi-label scenarios. In this paper, we propose the multi-label CircleSnake model for instance segmentation on Eos. It extends the original CircleSnake model from a single-label design to a multi-label model, allowing segmentation of multiple object types. Experimental results illustrate the CircleSnake model's superiority over the traditional Mask R-CNN model and DeepSnake model in terms of average precision (AP) in identifying and segmenting eosinophils, thereby enabling enhanced characterization of EoE. This automated approach holds promise for streamlining the assessment process and improving diagnostic accuracy in EoE analysis. The source code has been made publicly available at https://github.com/yilinliu610730/EoE.
Deep Learning-Based Open Source Toolkit for Eosinophil Detection in Pediatric Eosinophilic Esophagitis
Aug 11, 2023Eosinophilic Esophagitis (EoE) is a chronic, immune/antigen-mediated esophageal disease, characterized by symptoms related to esophageal dysfunction and histological evidence of eosinophil-dominant inflammation. Owing to the intricate microscopic representation of EoE in imaging, current methodologies which depend on manual identification are not only labor-intensive but also prone to inaccuracies. In this study, we develop an open-source toolkit, named Open-EoE, to perform end-to-end whole slide image (WSI) level eosinophil (Eos) detection using one line of command via Docker. Specifically, the toolkit supports three state-of-the-art deep learning-based object detection models. Furthermore, Open-EoE further optimizes the performance by implementing an ensemble learning strategy, and enhancing the precision and reliability of our results. The experimental results demonstrated that the Open-EoE toolkit can efficiently detect Eos on a testing set with 289 WSIs. At the widely accepted threshold of >= 15 Eos per high power field (HPF) for diagnosing EoE, the Open-EoE achieved an accuracy of 91%, showing decent consistency with pathologist evaluations. This suggests a promising avenue for integrating machine learning methodologies into the diagnostic process for EoE. The docker and source code has been made publicly available at https://github.com/hrlblab/Open-EoE.
Feasibility of Universal Anomaly Detection without Knowing the Abnormality in Medical Images
Jul 03, 2023



Many anomaly detection approaches, especially deep learning methods, have been recently developed to identify abnormal image morphology by only employing normal images during training. Unfortunately, many prior anomaly detection methods were optimized for a specific "known" abnormality (e.g., brain tumor, bone fraction, cell types). Moreover, even though only the normal images were used in the training process, the abnormal images were oftenly employed during the validation process (e.g., epoch selection, hyper-parameter tuning), which might leak the supposed ``unknown" abnormality unintentionally. In this study, we investigated these two essential aspects regarding universal anomaly detection in medical images by (1) comparing various anomaly detection methods across four medical datasets, (2) investigating the inevitable but often neglected issues on how to unbiasedly select the optimal anomaly detection model during the validation phase using only normal images, and (3) proposing a simple decision-level ensemble method to leverage the advantage of different kinds of anomaly detection without knowing the abnormality. The results of our experiments indicate that none of the evaluated methods consistently achieved the best performance across all datasets. Our proposed method enhanced the robustness of performance in general (average AUC 0.956).