 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Open-Source Knowledge Graph Ecosystem for the Life Sciences
Jul 11, 2023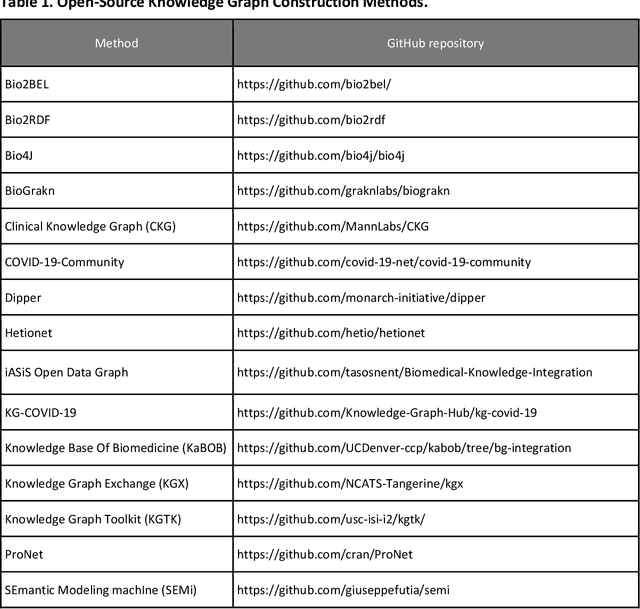
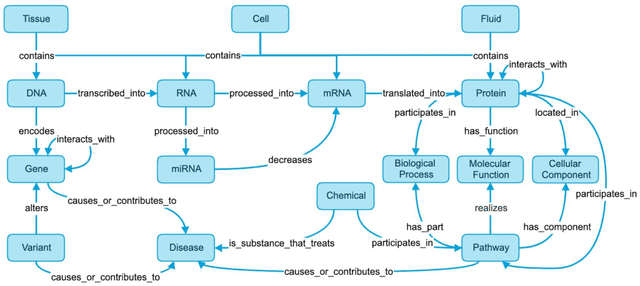
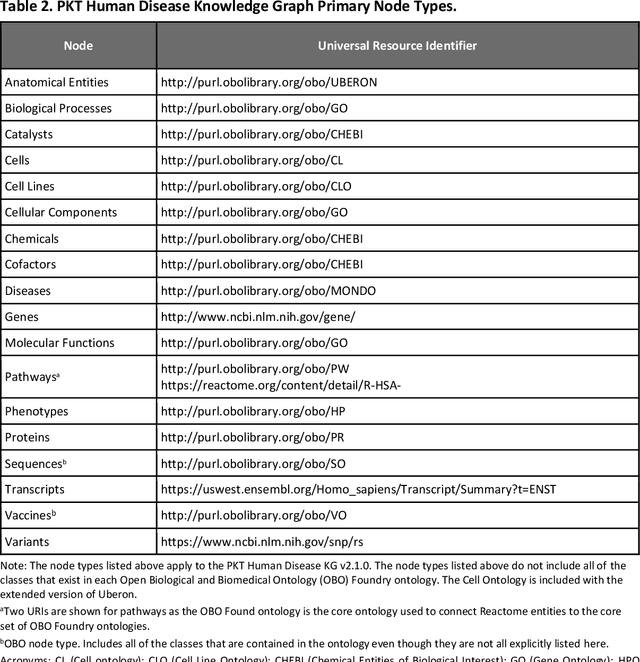
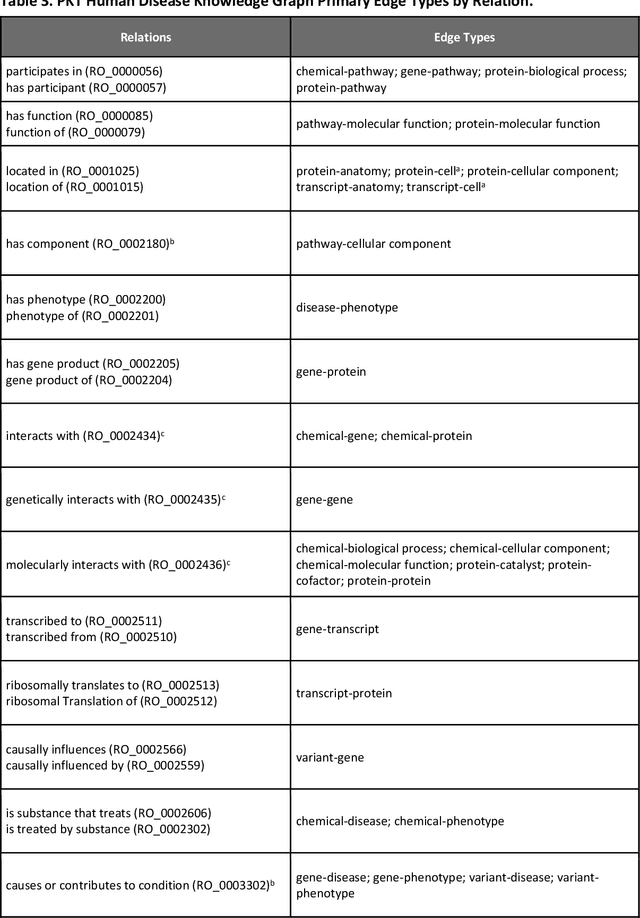
Translational research requires data at multiple scales of biological organization. Advancements in sequencing and multi-omics technologies have increased the availability of these data but researchers face significant integration challenges. Knowledge graphs (KGs) are used to model complex phenomena, and methods exist to automatically construct them. However, tackling complex biomedical integration problems requires flexibility in the way knowledge is modeled. Moreover, existing KG construction methods provide robust tooling at the cost of fixed or limited choices among knowledge representation models. PheKnowLator (Phenotype Knowledge Translator) is a semantic ecosystem for automating the FAIR (Findable, Accessible, Interoperable, and Reusable) construction of ontologically grounded KGs with fully customizable knowledge representation. The ecosystem includes KG construction resources (e.g., data preparation APIs), analysis tools (e.g., SPARQL endpoints and abstraction algorithms), and benchmarks (e.g., prebuilt KGs and embeddings). We evaluate the ecosystem by surveying open-source KG construction methods and analyzing its computational performance when constructing 12 large-scale KGs. With flexible knowledge representation, PheKnowLator enables fully customizable KGs without compromising performance or usability.
Structured prompt interrogation and recursive extraction of semantics (SPIRES): A method for populating knowledge bases using zero-shot learning
Apr 05, 2023
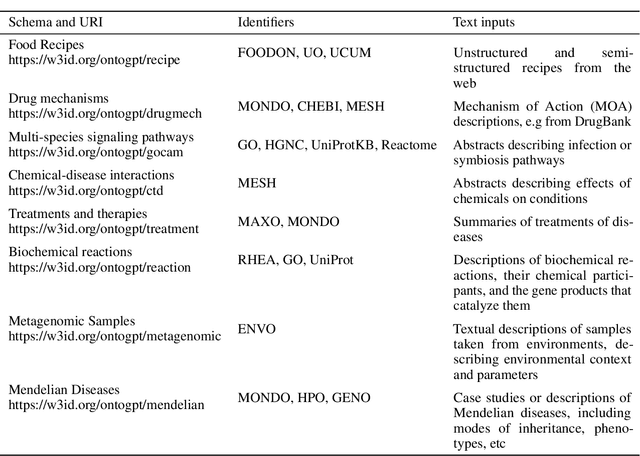
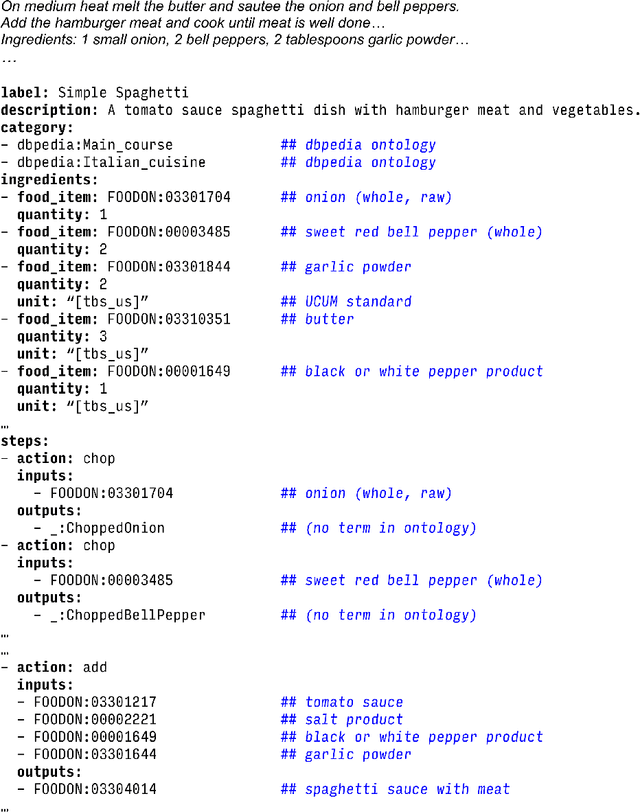

Creating knowledge bases and ontologies is a time consuming task that relies on a manual curation. AI/NLP approaches can assist expert curators in populating these knowledge bases, but current approaches rely on extensive training data, and are not able to populate arbitrary complex nested knowledge schemas. Here we present Structured Prompt Interrogation and Recursive Extraction of Semantics (SPIRES), a Knowledge Extraction approach that relies on the ability of Large Language Models (LLMs) to perform zero-shot learning (ZSL) and general-purpose query answering from flexible prompts and return information conforming to a specified schema. Given a detailed, user-defined knowledge schema and an input text, SPIRES recursively performs prompt interrogation against GPT-3+ to obtain a set of responses matching the provided schema. SPIRES uses existing ontologies and vocabularies to provide identifiers for all matched elements. We present examples of use of SPIRES in different domains, including extraction of food recipes, multi-species cellular signaling pathways, disease treatments, multi-step drug mechanisms, and chemical to disease causation graphs. Current SPIRES accuracy is comparable to the mid-range of existing Relation Extraction (RE) methods, but has the advantage of easy customization, flexibility, and, crucially, the ability to perform new tasks in the absence of any training data. This method supports a general strategy of leveraging the language interpreting capabilities of LLMs to assemble knowledge bases, assisting manual knowledge curation and acquisition while supporting validation with publicly-available databases and ontologies external to the LLM. SPIRES is available as part of the open source OntoGPT package: https://github.com/ monarch-initiative/ontogpt.
Ontologizing Health Systems Data at Scale: Making Translational Discovery a Reality
Sep 10, 2022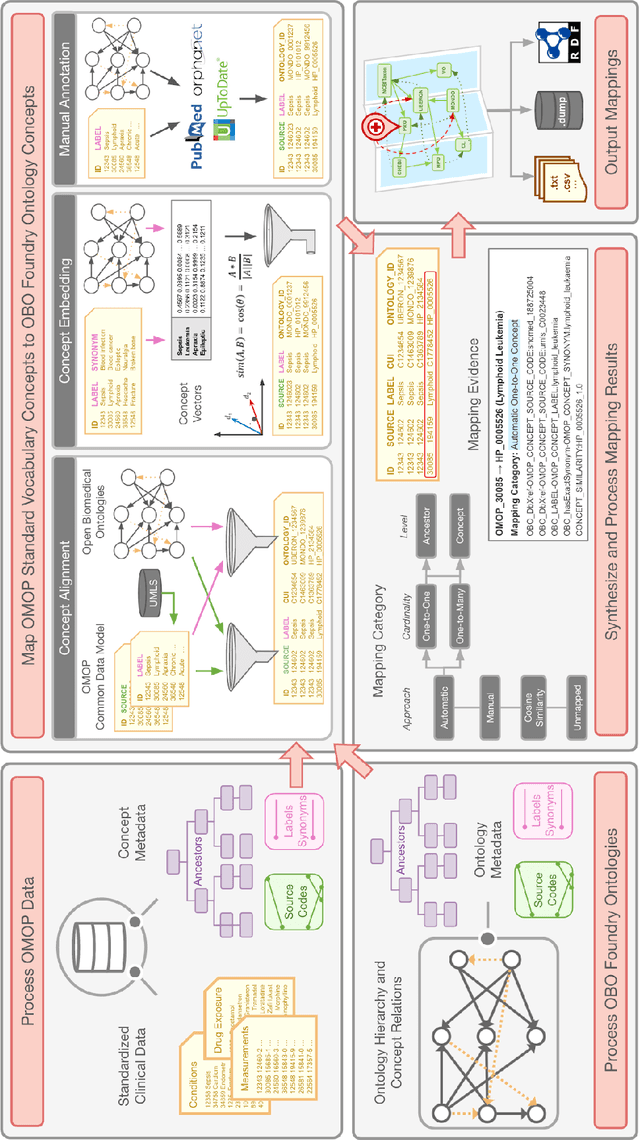
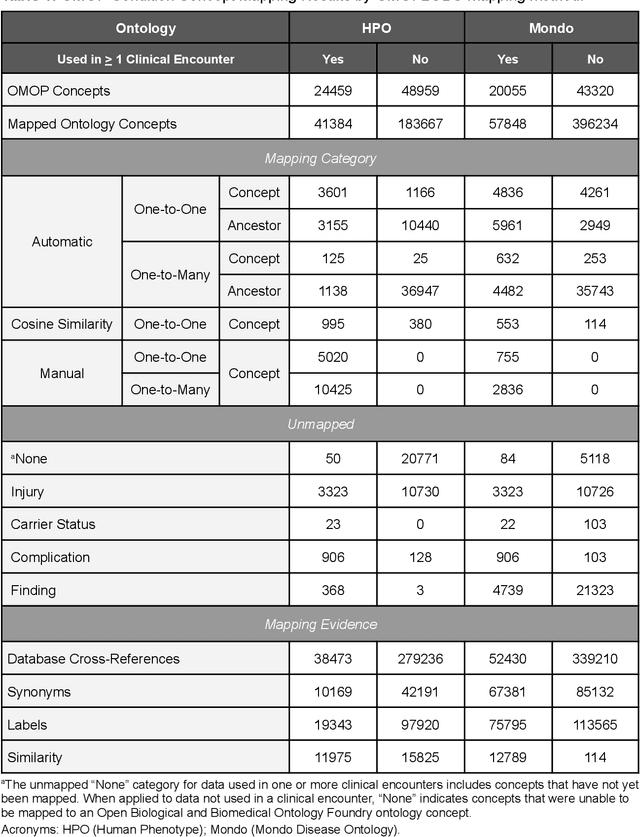
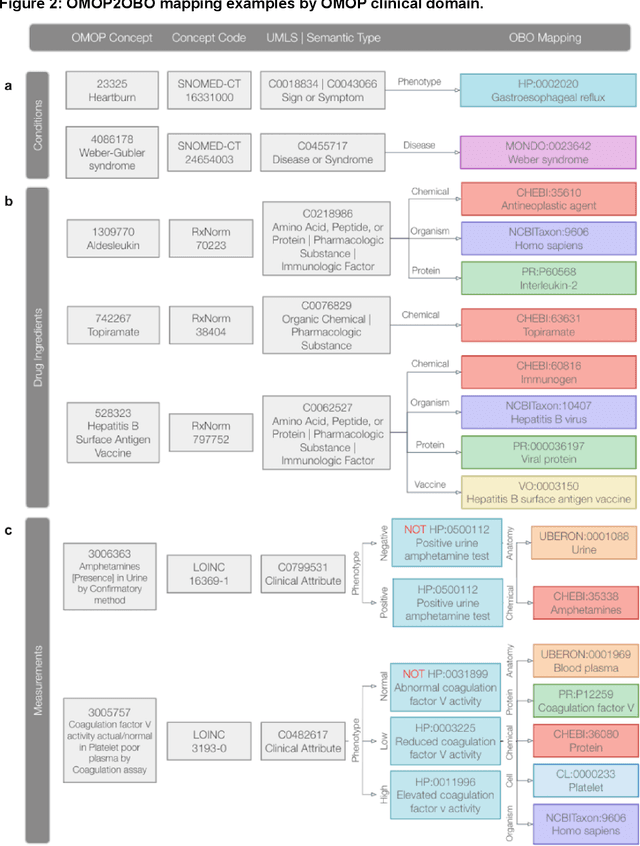
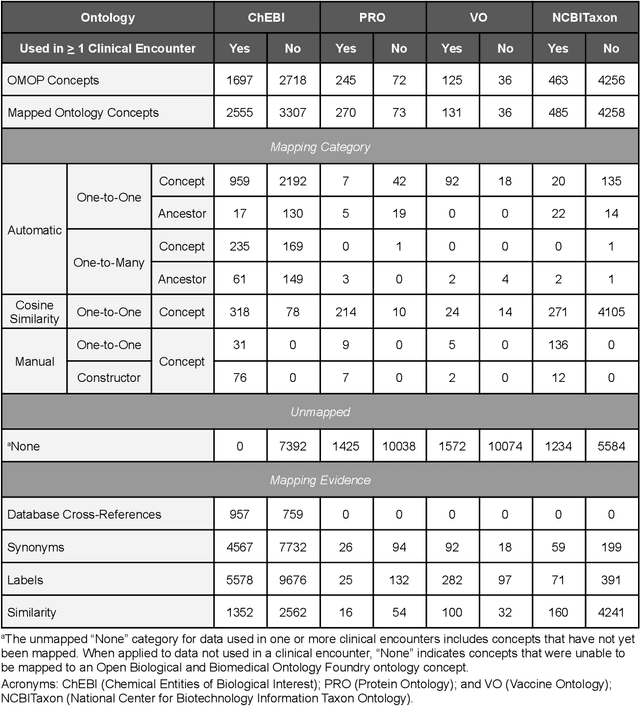
Common data models solve many challenges of standardizing electronic health record (EHR) data, but are unable to semantically integrate the resources needed for deep phenotyping. Open Biological and Biomedical Ontology (OBO) Foundry ontologies provide semantically computable representations of biological knowledge and enable the integration of a variety of biomedical data. However, mapping EHR data to OBO Foundry ontologies requires significant manual curation and domain expertise. We introduce a framework for mapping Observational Medical Outcomes Partnership (OMOP) standard vocabularies to OBO Foundry ontologies. Using this framework, we produced mappings for 92,367 conditions, 8,615 drug ingredients, and 10,673 measurement results. Mapping accuracy was verified by domain experts and when examined across 24 hospitals, the mappings covered 99% of conditions and drug ingredients and 68% of measurements. Finally, we demonstrate that OMOP2OBO mappings can aid in the systematic identification of undiagnosed rare disease patients who might benefit from genetic testing.
A Methodological Framework for the Comparative Evaluation of Multiple Imputation Methods: Multiple Imputation of Race, Ethnicity and Body Mass Index in the U.S. National COVID Cohort Collaborative
Jun 13, 2022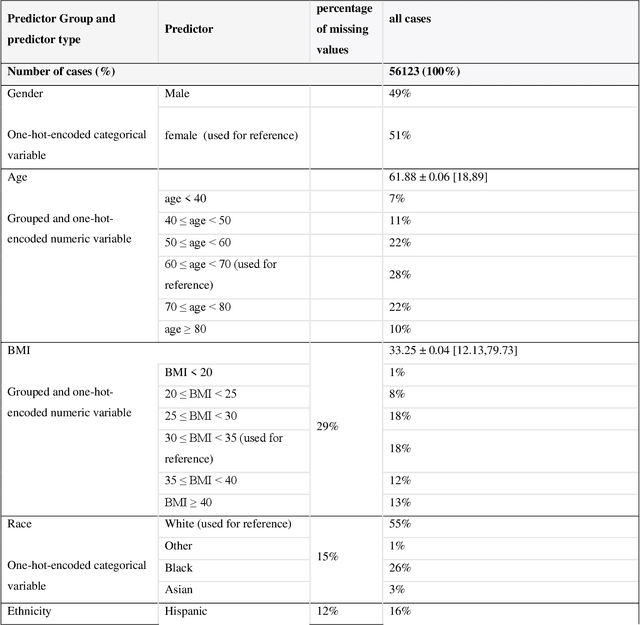
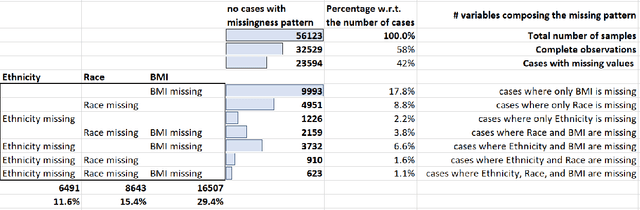

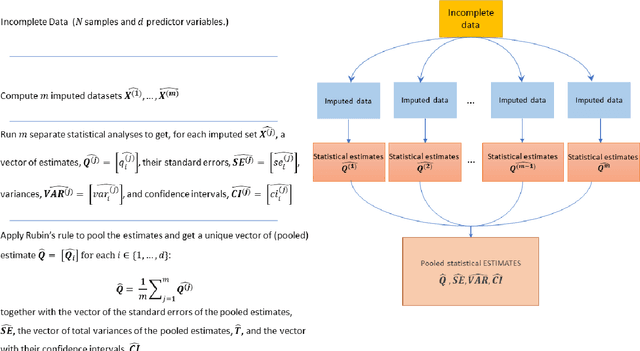
While electronic health records are a rich data source for biomedical research, these systems are not implemented uniformly across healthcare settings and significant data may be missing due to healthcare fragmentation and lack of interoperability between siloed electronic health records. Considering that the deletion of cases with missing data may introduce severe bias in the subsequent analysis, several authors prefer applying a multiple imputation strategy to recover the missing information. Unfortunately, although several literature works have documented promising results by using any of the different multiple imputation algorithms that are now freely available for research, there is no consensus on which MI algorithm works best. Beside the choice of the MI strategy, the choice of the imputation algorithm and its application settings are also both crucial and challenging. In this paper, inspired by the seminal works of Rubin and van Buuren, we propose a methodological framework that may be applied to evaluate and compare several multiple imputation techniques, with the aim to choose the most valid for computing inferences in a clinical research work. Our framework has been applied to validate, and extend on a larger cohort, the results we presented in a previous literature study, where we evaluated the influence of crucial patients' descriptors and COVID-19 severity in patients with type 2 diabetes mellitus whose data is provided by the National COVID Cohort Collaborative Enclave.
GraPE: fast and scalable Graph Processing and Embedding
Oct 12, 2021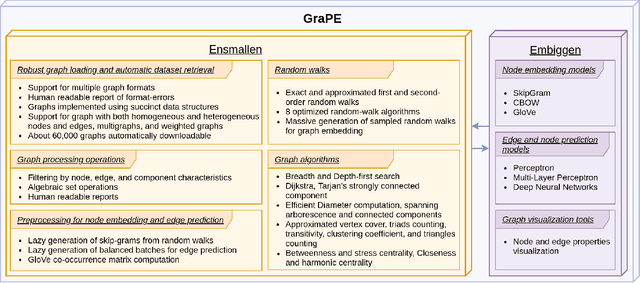
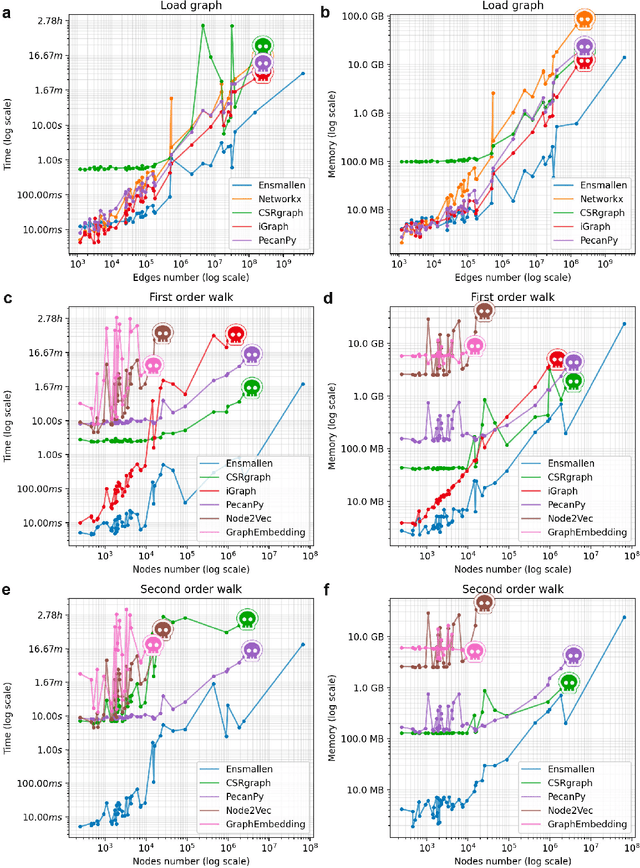
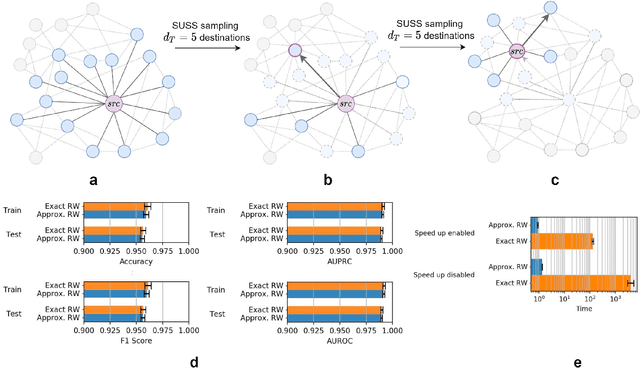
Graph Representation Learning methods have enabled a wide range of learning problems to be addressed for data that can be represented in graph form. Nevertheless, several real world problems in economy, biology, medicine and other fields raised relevant scaling problems with existing methods and their software implementation, due to the size of real world graphs characterized by millions of nodes and billions of edges. We present GraPE, a software resource for graph processing and random walk based embedding, that can scale with large and high-degree graphs and significantly speed up-computation. GraPE comprises specialized data structures, algorithms, and a fast parallel implementation that displays everal orders of magnitude improvement in empirical space and time complexity compared to state of the art software resources, with a corresponding boost in the performance of machine learning methods for edge and node label prediction and for the unsupervised analysis of graphs.GraPE is designed to run on laptop and desktop computers, as well as on high performance computing clusters
PhenoTagger: A Hybrid Method for Phenotype Concept Recognition using Human Phenotype Ontology
Sep 17, 2020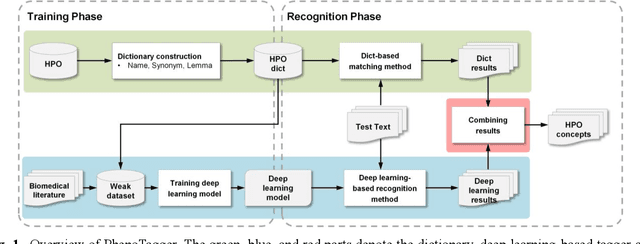
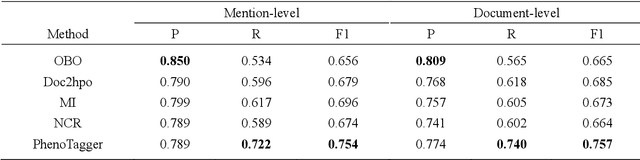
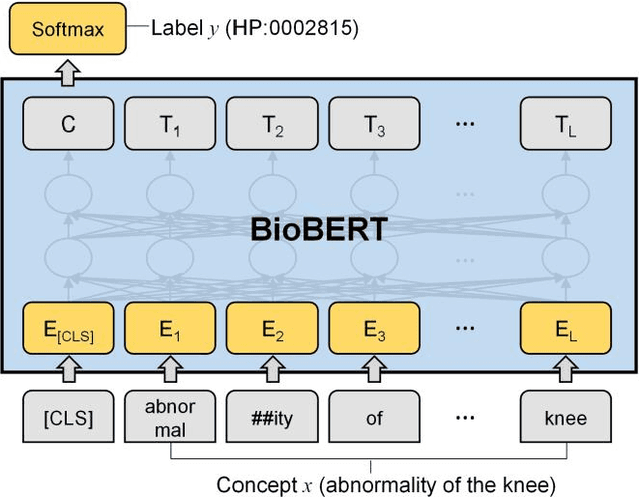
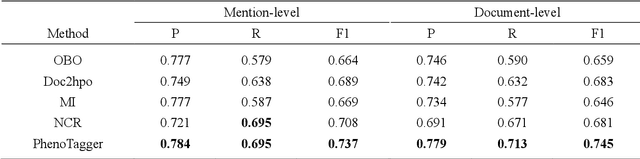
Automatic phenotype concept recognition from unstructured text remains a challenging task in biomedical text mining research. Previous works that address the task typically use dictionary-based matching methods, which can achieve high precision but suffer from lower recall. Recently, machine learning-based methods have been proposed to identify biomedical concepts, which can recognize more unseen concept synonyms by automatic feature learning. However, most methods require large corpora of manually annotated data for model training, which is difficult to obtain due to the high cost of human annotation. In this paper, we propose PhenoTagger, a hybrid method that combines both dictionary and machine learning-based methods to recognize Human Phenotype Ontology (HPO) concepts in unstructured biomedical text. We first use all concepts and synonyms in HPO to construct a dictionary. Then, the dictionary and biomedical literature are used to automatically build a weakly-supervised training dataset for machine learning. Next, a cutting-edge deep learning model is trained to classify each candidate phrase into a corresponding concept label. Finally, the dictionary and machine learning-based prediction results are combined for improved performance. Our method is validated with two HPO corpora, and the results show that PhenoTagger compares favorably to state-of-the-art methods. In addition, to demonstrate the generalizability of our method, we retrained PhenoTagger using the disease ontology MEDIC for disease concept recognition to investigate the effect of training on different ontologies. Experimental results on the NCBI disease corpus show that PhenoTagger without requiring manually annotated training data achieves competitive performance as compared with state-of-the-art supervised methods.