 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemical classification program synthesis using generative artificial intelligence
May 24, 2025Accurately classifying chemical structures is essential for cheminformatics and bioinformatics, including tasks such as identifying bioactive compounds of interest, screening molecules for toxicity to humans, finding non-organic compounds with desirable material properties, or organizing large chemical libraries for drug discovery or environmental monitoring. However, manual classification is labor-intensive and difficult to scale to large chemical databases. Existing automated approaches either rely on manually constructed classification rules, or the use of deep learning methods that lack explainability. This work presents an approach that uses generative artificial intelligence to automatically write chemical classifier programs for classes in the Chemical Entities of Biological Interest (ChEBI) database. These programs can be used for efficient deterministic run-time classification of SMILES structures, with natural language explanations. The programs themselves constitute an explainable computable ontological model of chemical class nomenclature, which we call the ChEBI Chemical Class Program Ontology (C3PO). We validated our approach against the ChEBI database, and compared our results against state of the art deep learning models. We also demonstrate the use of C3PO to classify out-of-distribution examples taken from metabolomics repositories and natural product databases. We also demonstrate the potential use of our approach to find systematic classification errors in existing chemical databases, and show how an ensemble artificial intelligence approach combining generated ontologies, automated literature search, and multimodal vision models can be used to pinpoint potential errors requiring expert validation
The Artificial Intelligence Ontology: LLM-assisted construction of AI concept hierarchies
Apr 03, 2024


The Artificial Intelligence Ontology (AIO) is a systematization of artificial intelligence (AI) concepts, methodologies, and their interrelations. Developed via manual curation, with the additional assistance of large language models (LLMs), AIO aims to address the rapidly evolving landscape of AI by providing a comprehensive framework that encompasses both technical and ethical aspects of AI technologies. The primary audience for AIO includes AI researchers, developers, and educators seeking standardized terminology and concepts within the AI domain. The ontology is structured around six top-level branches: Networks, Layers, Functions, LLMs, Preprocessing, and Bias, each designed to support the modular composition of AI methods and facilitate a deeper understanding of deep learning architectures and ethical considerations in AI. AIO's development utilized the Ontology Development Kit (ODK) for its creation and maintenance, with its content being dynamically updated through AI-driven curation support. This approach not only ensures the ontology's relevance amidst the fast-paced advancements in AI but also significantly enhances its utility for researchers, developers, and educators by simplifying the integration of new AI concepts and methodologies. The ontology's utility is demonstrated through the annotation of AI methods data in a catalog of AI research publications and the integration into the BioPortal ontology resource, highlighting its potential for cross-disciplinary research. The AIO ontology is open source and is available on GitHub (https://github.com/berkeleybop/artificial-intelligence-ontology) and BioPortal (https://bioportal.bioontology.org/ontologies/AIO).
Automated Annotation of Scientific Texts for ML-based Keyphrase Extraction and Validation
Nov 08, 2023Advanced omics technologies and facilities generate a wealth of valuable data daily; however, the data often lacks the essential metadata required for researchers to find and search them effectively. The lack of metadata poses a significant challenge in the utilization of these datasets. Machine learning-based metadata extraction techniques have emerged as a potentially viable approach to automatically annotating scientific datasets with the metadata necessary for enabling effective search. Text labeling, usually performed manually, plays a crucial role in validating machine-extracted metadata. However, manual labeling is time-consuming; thus, there is an need to develop automated text labeling techniques in order to accelerate the process of scientific innovation. This need is particularly urgent in fields such as environmental genomics and microbiome science, which have historically received less attention in terms of metadata curation and creation of gold-standard text mining datasets. In this paper, we present two novel automated text labeling approaches for the validation of ML-generated metadata for unlabeled texts, with specific applications in environmental genomics. Our techniques show the potential of two new ways to leverage existing information about the unlabeled texts and the scientific domain. The first technique exploits relationships between different types of data sources related to the same research study, such as publications and proposals. The second technique takes advantage of domain-specific controlled vocabularies or ontologies. In this paper, we detail applying these approaches for ML-generated metadata validation. Our results show that the proposed label assignment approaches can generate both generic and highly-specific text labels for the unlabeled texts, with up to 44% of the labels matching with those suggested by a ML keyword extraction algorithm.
MapperGPT: Large Language Models for Linking and Mapping Entities
Oct 05, 2023Aligning terminological resources, including ontologies, controlled vocabularies, taxonomies, and value sets is a critical part of data integration in many domains such as healthcare, chemistry, and biomedical research. Entity mapping is the process of determining correspondences between entities across these resources, such as gene identifiers, disease concepts, or chemical entity identifiers. Many tools have been developed to compute such mappings based on common structural features and lexical information such as labels and synonyms. Lexical approaches in particular often provide very high recall, but low precision, due to lexical ambiguity. As a consequence of this, mapping efforts often resort to a labor intensive manual mapping refinement through a human curator. Large Language Models (LLMs), such as the ones employed by ChatGPT, have generalizable abilities to perform a wide range of tasks, including question-answering and information extraction. Here we present MapperGPT, an approach that uses LLMs to review and refine mapping relationships as a post-processing step, in concert with existing high-recall methods that are based on lexical and structural heuristics. We evaluated MapperGPT on a series of alignment tasks from different domains, including anatomy, developmental biology, and renal diseases. We devised a collection of tasks that are designed to be particularly challenging for lexical methods. We show that when used in combination with high-recall methods, MapperGPT can provide a substantial improvement in accuracy, beating state-of-the-art (SOTA) methods such as LogMap.
An Open-Source Knowledge Graph Ecosystem for the Life Sciences
Jul 11, 2023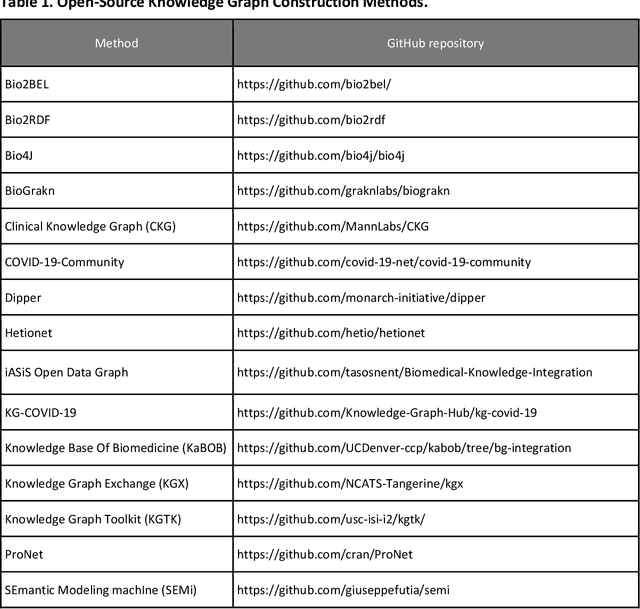
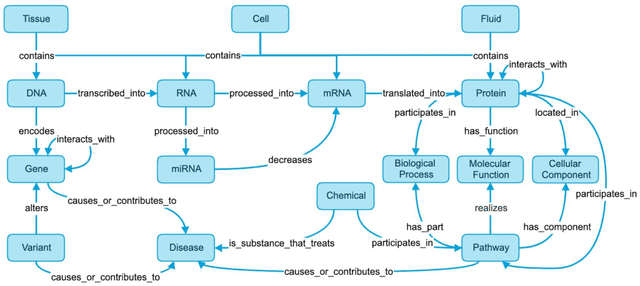
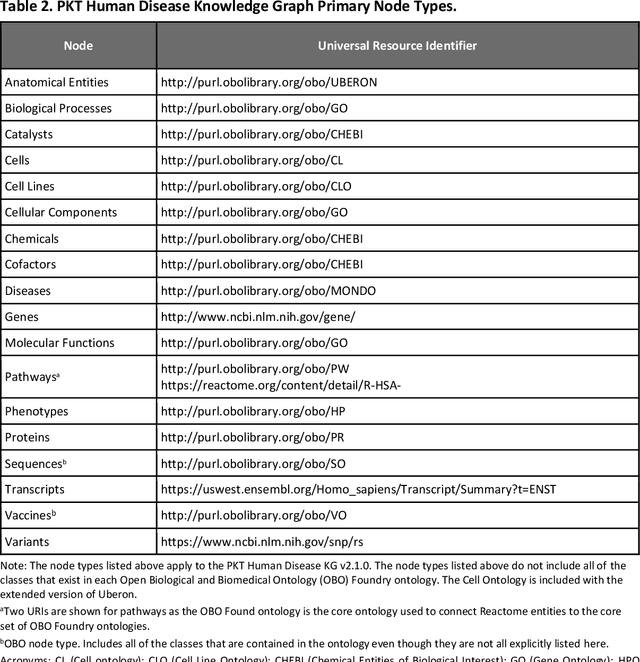
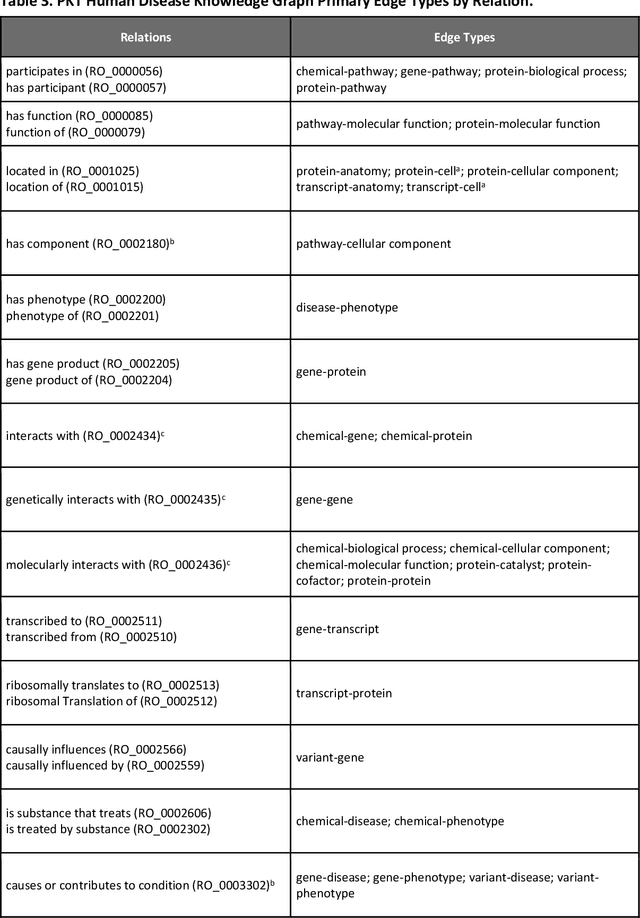
Translational research requires data at multiple scales of biological organization. Advancements in sequencing and multi-omics technologies have increased the availability of these data but researchers face significant integration challenges. Knowledge graphs (KGs) are used to model complex phenomena, and methods exist to automatically construct them. However, tackling complex biomedical integration problems requires flexibility in the way knowledge is modeled. Moreover, existing KG construction methods provide robust tooling at the cost of fixed or limited choices among knowledge representation models. PheKnowLator (Phenotype Knowledge Translator) is a semantic ecosystem for automating the FAIR (Findable, Accessible, Interoperable, and Reusable) construction of ontologically grounded KGs with fully customizable knowledge representation. The ecosystem includes KG construction resources (e.g., data preparation APIs), analysis tools (e.g., SPARQL endpoints and abstraction algorithms), and benchmarks (e.g., prebuilt KGs and embeddings). We evaluate the ecosystem by surveying open-source KG construction methods and analyzing its computational performance when constructing 12 large-scale KGs. With flexible knowledge representation, PheKnowLator enables fully customizable KGs without compromising performance or usability.
Gene Set Summarization using Large Language Models
May 25, 2023Molecular biologists frequently interpret gene lists derived from high-throughput experiments and computational analysis. This is typically done as a statistical enrichment analysis that measures the over- or under-representation of biological function terms associated with genes or their properties, based on curated assertions from a knowledge base (KB) such as the Gene Ontology (GO). Interpreting gene lists can also be framed as a textual summarization task, enabling the use of Large Language Models (LLMs), potentially utilizing scientific texts directly and avoiding reliance on a KB. We developed SPINDOCTOR (Structured Prompt Interpolation of Natural Language Descriptions of Controlled Terms for Ontology Reporting), a method that uses GPT models to perform gene set function summarization as a complement to standard enrichment analysis. This method can use different sources of gene functional information: (1) structured text derived from curated ontological KB annotations, (2) ontology-free narrative gene summaries, or (3) direct model retrieval. We demonstrate that these methods are able to generate plausible and biologically valid summary GO term lists for gene sets. However, GPT-based approaches are unable to deliver reliable scores or p-values and often return terms that are not statistically significant. Crucially, these methods were rarely able to recapitulate the most precise and informative term from standard enrichment, likely due to an inability to generalize and reason using an ontology. Results are highly nondeterministic, with minor variations in prompt resulting in radically different term lists. Our results show that at this point, LLM-based methods are unsuitable as a replacement for standard term enrichment analysis and that manual curation of ontological assertions remains necessary.
Structured prompt interrogation and recursive extraction of semantics (SPIRES): A method for populating knowledge bases using zero-shot learning
Apr 05, 2023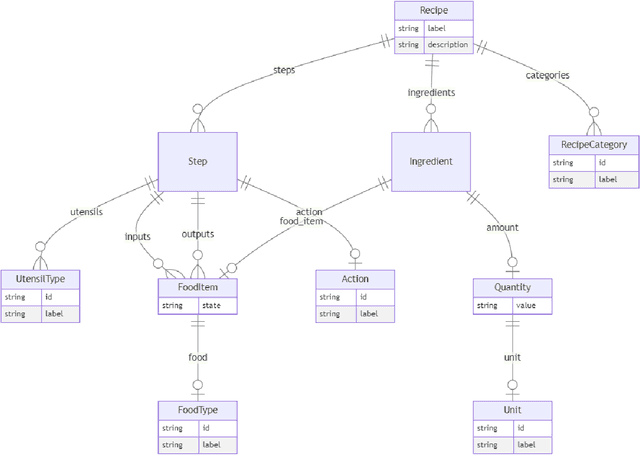
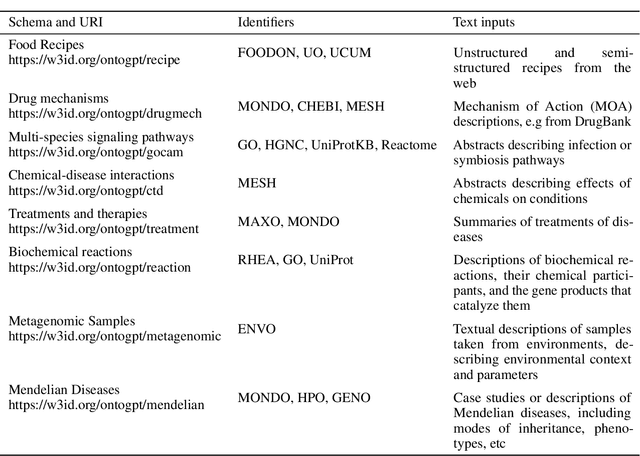
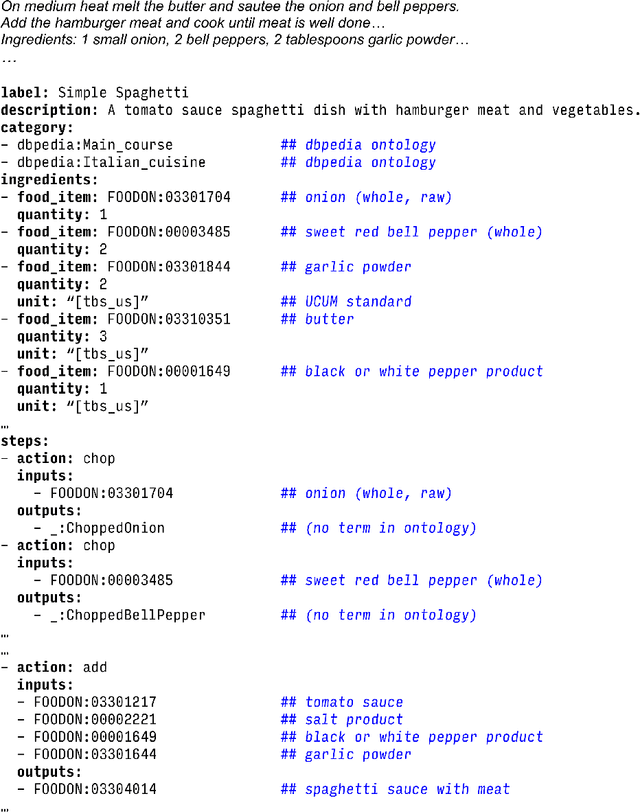

Creating knowledge bases and ontologies is a time consuming task that relies on a manual curation. AI/NLP approaches can assist expert curators in populating these knowledge bases, but current approaches rely on extensive training data, and are not able to populate arbitrary complex nested knowledge schemas. Here we present Structured Prompt Interrogation and Recursive Extraction of Semantics (SPIRES), a Knowledge Extraction approach that relies on the ability of Large Language Models (LLMs) to perform zero-shot learning (ZSL) and general-purpose query answering from flexible prompts and return information conforming to a specified schema. Given a detailed, user-defined knowledge schema and an input text, SPIRES recursively performs prompt interrogation against GPT-3+ to obtain a set of responses matching the provided schema. SPIRES uses existing ontologies and vocabularies to provide identifiers for all matched elements. We present examples of use of SPIRES in different domains, including extraction of food recipes, multi-species cellular signaling pathways, disease treatments, multi-step drug mechanisms, and chemical to disease causation graphs. Current SPIRES accuracy is comparable to the mid-range of existing Relation Extraction (RE) methods, but has the advantage of easy customization, flexibility, and, crucially, the ability to perform new tasks in the absence of any training data. This method supports a general strategy of leveraging the language interpreting capabilities of LLMs to assemble knowledge bases, assisting manual knowledge curation and acquisition while supporting validation with publicly-available databases and ontologies external to the LLM. SPIRES is available as part of the open source OntoGPT package: https://github.com/ monarch-initiative/ontogpt.
GraPE: fast and scalable Graph Processing and Embedding
Oct 12, 2021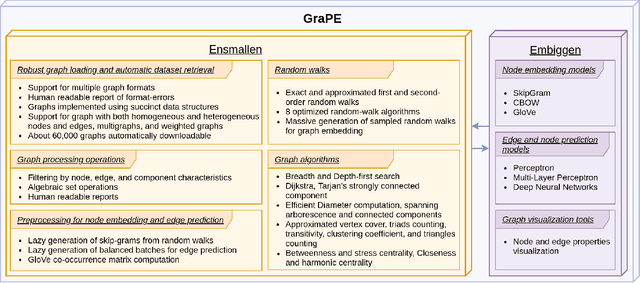
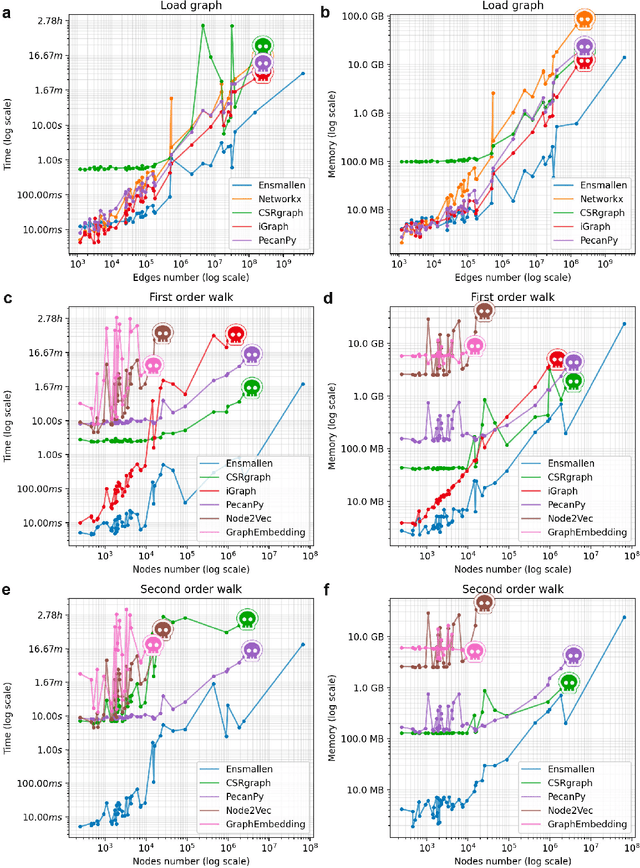
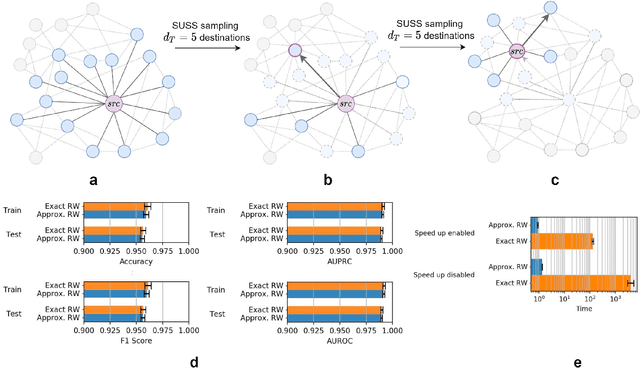
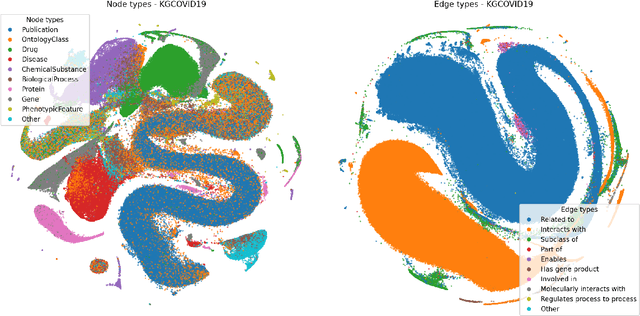
Graph Representation Learning methods have enabled a wide range of learning problems to be addressed for data that can be represented in graph form. Nevertheless, several real world problems in economy, biology, medicine and other fields raised relevant scaling problems with existing methods and their software implementation, due to the size of real world graphs characterized by millions of nodes and billions of edges. We present GraPE, a software resource for graph processing and random walk based embedding, that can scale with large and high-degree graphs and significantly speed up-computation. GraPE comprises specialized data structures, algorithms, and a fast parallel implementation that displays everal orders of magnitude improvement in empirical space and time complexity compared to state of the art software resources, with a corresponding boost in the performance of machine learning methods for edge and node label prediction and for the unsupervised analysis of graphs.GraPE is designed to run on laptop and desktop computers, as well as on high performance computing clusters